
In the beginning, scientific articles were just letters. Scholars wrote to each other about whatever they were working on, celebrating their discoveries or arguing over minutiae, and ended up with great stacks of the things. People started bringing interesting letters to meetings of the Royal Society to read aloud, then scientists started addressing their letters to the Royal Society directly, and eventually Henry Oldenburg started pulling some of these letters together and printing them as the Philosophical Transactions of the Royal Society, the first scientific journal.

In continuance of this hallowed tradition, in this blog post we are publishing some philosophical transactions of our own: correspondence with JP Callaghan, an MD/PhD student at a large Northeast research university going into anesthesia. He has expertise in protein statistical mechanics and kinetic modeling, so he reached out to us with several ideas and enlightened criticisms.
With JP Callaghan’s help we have lightly edited the correspondence for clarity, turning the multi-threaded format of the email exchange into something more linear. We found the conversation very informative, and we hope you do as well! So without further ado:
JP Callaghan: Hi guys, great work on A Chemical Hunger.
I’m sure someone already suggested this but the Fulbright program executes the “move abroad” experiment every year. In fact, they do the reverse experiment as well, paying foreigners to move to the US. The Phillipines Fulbright program seems especially active.
(The Peace Corps is already doing this experiment as well, but that’s probably probably more confounded since people are often living in pretty rustic locations.)
You could pretty easily imagine paying these folks a little extra money to send you their weight once a month or whatever.
SLIME MOLD TIME MOLD: Thank you! Yeah, we’ve been trying to figure out the best way to pursue this one, using existing data if possible. Fulbright is a good idea, especially US <–––> Philippines, and especially because we suspect young people will show weight changes faster. We’ve also thought about trying to collect a sample of expats, possibly on reddit, since there are a lot of anecdotes of weight loss in those communities.
The tricky thing is finding someone who has an in with one of these groups. We probably can’t just cold call Fulbright and ask how much all their scholars weigh, though we’ll start asking around.
JPC: Unfortunately my connection with the Fulbright was brief, superficial, and many years ago. I can ask around at my university, though. I’m not filled with unmitigated optimism, but the worst they can do is say no/ignore me.
Also, I wanted to mention that lithium level measurements are extremely common measurements in clinical practice. It’s used to monitor therapeutic lithium (for e.g. bipolar folks). (Although I will concede usually they are measuring .5 – 1.5 mmol/L which would be way higher than serum levels due to contamination.) Also, it’s interesting that the early pharmacokinetic studies also measured urine lithium (see e.g. Barbara Ehrlich’s seminal 1980 paper) so there’s precedent for that as well. I’m led to understand from my lab medicine colleagues that it’s a relatively straightforward (aka cheap) electrochemical assay, at least in common clinical practice.
SMTM: We’ve looked into measurement a bit. We’re concerned that serum levels aren’t worth measuring, since lithium seems to accumulate in the brain and we suspect that would be the mechanism (a commenter suggested it might also be accumulation in bone). But if we were to do clinical measurements, we’d probably measure lithium in urine or maybe even in saliva, since there’s evidence they’re good proxies for one another and for the levels in serum, and they’re easier to collect. Urine might be especially important if lithium clearance rate ends up being a piece of the puzzle, which it seems like it might.
JPC: It is definitely true that lithium accumulates inside cells (definitely rat neurons and human RBCs, probably human neurons, but maybe not human muscle; see e.g. that Ehrlich paper I mentioned). The thing is, lithium kinetics seem to be pretty fast. Since it’s an ion, it doesn’t partition into fat the way other long-lasting medications and toxins do, and so it’s eliminated fairly quickly by the kidneys. (THC is a classic example of a hydrophobic “contaminant”; this same physical chemistry explains why a long-time pothead will test positive for THC for months, but you can stop using cocaine and, 72 hours later, screen negative.)
It might be worth your time to look at some of the lithium washout experiments that have been done over the years (e.g. Hunter, 1988 where they see lithium levels rapidly decline after stopping lithium therapy that had been going on for a month).
I suppose, though, that I’m not aware of any data that specifically excludes the possibility that there is a very slow “third compartment” where lithium can deposit (such as, as your commenter suggested, bone; although I don’t know much about whether or not lithium can incorporate into the hydroxyapatite matrix in bone. It’s mostly calcium phosphate and I’m not sure if lithium could “find a place” in that crystalline matrix).
Anyway, though, my understanding is that lithium kinetics in the brain are relatively fast. (For instance, see Ebadi, et al where they measure [Li] in rat brains over time.) So even if you have a highly accumulated slow bone compartment, the levels of lithium you’d get in the brain would still be super low, because it equilibrates with the blood quickly and therefore is subject to rapid elimination by the kidneys.
However, I don’t think you need to posit accumulation for your hypothesis. If you’re exposed to constant, low levels of lithium, you reach an equilibrium. There’s some super low serum concentration, some rather-higher intracellular concentration, and it’s all held in steady state by the constant intake via the GI tract (say, in the water) and constant elimination by the kidneys. Perhaps this is what you’re getting at when you say the rate of elimination might be very important?
Instead, consider some interesting pharmacodynamics: low-level (or maybe widely fluctuating, since lithium is also quickly cleared?) exposure to lithium messes with the lipostat. This process is probably really slow, maybe because weight change is slow or maybe because of some kind of brain adaptation process or whatever. We have good reason to suspect low-level lithium has neurological effects already anyway through some of the population-level suicide data I’m sure you’re aware of.
Urine and serum levels of lithium are only good proxies for one another at steady state. I really strongly suggest you guys look at that Ehrlich paper. She measures serum, intra-RBC, and urine [Li] after a dose of lithium carbonate (the most common delayed-release preparation of pharmaceutical lithium).
Another good one is Gaillot et al which demonstrates how important the form of lithium (lithium carbonate vs LiCl) is to the kinetics. (As an aside, this might be a reason for lithium grease to be so bad; lithium grease is apparently some kind of weird soap complex with fatty acids, maybe it gets trapped in the GI tract or something.)
SMTM: The rat studies are interesting but don’t rats seem like a bad comparison for determining something like rate of clearance? Besides just not being human, their metabolisms are something like 6-8x faster than ours and their lifespans are about 20 times shorter. Also human brains are huge. What do you think?
JPC: Certainly I agree that rats are not people and are bad models in many ways. I think that renal function is the key parameter you’d want to compare. The most basic measure of kidney function is the GFR (glomerular filtration rate), which basically measures how much fluid gets pushed through the “kidney filter” per unit time. Unfortunately in people we measure it in volume/time/body surface area and in rats volume/time/mass which makes a comparison less obvious than I was hoping. To be honest, I am not sure how well rat kidney function and human kidney function is comparable. (Definitely more comparable than live and dead human kidney function, though .)
What do you mean by ”their metabolisms are something like 6-8x faster than ours”? Like, calories/mass/time? Usually when I think about “metabolic rate” I am thinking of energy usage. When we think about drug elimination, the main things that matter are 1) liver function (for drugs that are hepatically metabolized) 2) various tissue enzyme function (e.g. plasma esterases for something like esmolol) and 3) renal function. I don’t generally think about basal metabolic rate as being a pertinent factor, really, except perhaps in cases where it’s a proxy for hepatic metabolism.
Lithium is eliminated (“cleared”) almost exclusively by the kidney and it undergoes no metabolic transformations, so I wouldn’t worry about anything but kidney function for its clearance.
You’re right, though, the 20x lifespan difference could be an issue. If we are worried about accumulation on the timescale of years, then obviously a shorter rat life is a problem. But (if I read your blog posts right) rats as experimental animals are also getting fatter so presumably the effect extends to them on the timescale of their life? (Did you have data in rats? I don’t remember.)
Indeed, if it’s actually just that there a constant low-level “infusion” of lithium via tapwater, grease exposure at work, etc giving rise to a low steady-state lithium (rather than actual bioaccumulation) this would explain why the effect does extend to these short-lived experimental animals.
SMTM: You make good points about laboratory animals. There are data on rats and they do seem to be getting heavier. Let’s stick a pin in this one for a now, you may find this next bit is relevant to the same questions:
In your opinion, are the studies you cite consistent or inconsistent with the findings of Amdisen et al. 1974 and Shoepfer et al. 2021? Also potentially relevant is Amidsen 1977. We describe their findings near the end of this section — basically they seem to suggest that Li accumulates preferentially in the bones, thyroid, and parts of the brain. The total sample size is small but it seems suggestive. We agree accumulation may not be essential to the theory but doesn’t this look like evidence of accumulation? We’ve attached copies of Amdisen et al. 1974 and Amdisen 1977 as PDFs in case you want to take a closer look. [SMTM’s Note: If anyone else wants to see these papers, you can email us.]
Especially interesting that Ebadi et al. say, “it has been shown that sodium intake exerts a significant influence on the renal elimination of lithium (Schou, 1958b)”, somewhat in line with our speculation here. We’ll have to look into that.
Brains
JPC: Thanks for the papers. As you predicted, I’m finding them super interesting.
Shoepfer et al, 2021 is a lovely, very interesting paper (complete with some adorable Deutsch-English). I was aware of it but had not taken the time to read it yet.
By my read, it is primarily seeking to establish this new, nuclear fission based approach to measuring lithium in pathology tissue. After spending some time with it, I don’t really know how to interpret their findings. The main reason I am not sure what to do with this paper is that the results are in dead peoples’ brains. Indeed, they specifically note in their ‘limitations’ section: “The lithium distribution patterns so far obtained with the NIK method, thus in no way contradicting given literature references, are based on post mortem tissue.” The reason this is pertinent is that there is a lot of active transport of other monovalent cations (K, Na) and so I would worry that this is true for lithium as well and (obviously) this is almost certainly disrupted in dead people.
The second thing is that the tissue was fixed in (presumably) formalin and stained with hematoxylin and eosin before measuring lithium, which then comes out in units of mass/mass. Obviously in living tissue there’s lots of water and whatnot, and the mass-density of water and formalin is going to be pretty different.
So, as the authors say, I would say it’s neither consistent nor inconsistent with other data.
SMTM: It’s true that all the brain samples we have in humans are in dead brain tissue, but this seems like an insurmountable issue, right? Looking at dead tissue is the only way to get even a rough estimate of how much lithium is in the brain, since as far as we know there’s no way to test the levels in a living human brain, or if there is, no one has taken those measurements and it’s outside our current budget.
In any case, the most relevant findings from these studies, at least in our opinion, are 1) that lithium definitely reaches brain tissue and sticks around for a while, and 2) regardless of absolute levels, there seems to be relatively more lithium in parts of the brain that regulate appetite and weight gain. These conclusions seem likely to hold even given all the reasonable concerns about dead tissue. What do you think?
JPC: I agree. In my mind, the main question is whether or not lithium persists in the brain after cessation of lithium therapy. Put more rigorously, what is the rate of exchange between the “brain compartment” and (probably) the “serum compartment.” (I guess it could also be eliminated by CSF too maybe? Or “glymphatics”? idk I guess nobody really understands the brain.)

The main issue I have is this: if you’re exposed, say, to 20 ppb lithium and your serum has 20 ppb lithium and so does the cytoplasm in your neurons, this is actually the null hypothesis (that lithium is an inert substance that just flows down its concentration gradient). It’s obviously false (we know lithium concentrates in RBCs of healthy subjects, for instance), but this paper doesn’t help me decide if lithium 1) passively diffuses throughout the body 2) is actively concentrated in neurons, or even 3) is actively cleared from cells, simply because I don’t really know what to do with the number.
The second issue is the preparation. Maybe formalin fixation washes lithium away, or when it fixes cell membranes maybe the lithium is allowed to diffuse out. Maybe it poorly penetrates myelin sheaths, and has a tendency to concentrate the lithium inside cells by making the extracellular environment more hydrophobic (nature abhors an unsolvated ion).
Another reason I am so skeptical of the “slow lithium kinetics” hypothesis is just the physical chemistry of lithium. It’s a tiny, charged particle. Keeping these sorts of ions from moving around and distributing evenly is actually really hard in most cases. There are a few cases of ionic solids in the human body (various types of kidney stones, bones, bile stones] but for the most part these involve much less soluble ions than lithium and everything is dissolved and flows around at its whim except where it’s actively pumped.
SMTM: This is a good point, and in addition, the fact that tourists and expats seem to lose weight quickly does seem to be a point in favor of fast lithium over slow lithium. If those anecdotes bear out in some kind of more systematic study, “slow lithium kinetics” starts looking really unlikely. Another possibility, though, is that young people are the only ones who lose weight quickly on foreign trips, and there’s something like a “weight gain in the brain, reservoir in the bone” system where people remain dosed for a long time once enough has built up in their bones (or some other reservoir).
JPC: Very possible. Also young people generally have better renal function. There are tons of people walking around with their kidneys at like 50% or worse who don’t even know it.
A third and distant issue what I mentioned about the active transport of Na and K that happens in neurons (IIRC something like 1/3 of your calories are spent doing this) ceasing when you’re dead. This is also a fairly big deal, though, since there are various cation leak channels in cell membranes (for electrical excitability reasons, I think; ask an electrical engineer or a different kind of biophysicist) through which Li might also escape. (Since, after all, a reasonable hypothesis for the mechanism of action is that Li uses Na channels.)
Between these three difficulties, I do actually see this as borderline insurmountable for ascertaining how much lithium is in an alive brain based on these data. Basically, it comes down to “I don’t know how much lithium I should expect there to be in these experiments.”
However, “relatively more lithium in parts of the brain that regulate appetite and weight gain” is a good point. I think that this is something you actually can reasonably say: it seems like there is more lithium in these areas than other areas. The within-experiment comparisons definitely seem more sound. It would also be consistent with the onset of hunger/appetite symptoms below traditionally-accepted therapeutic ranges.
I do also want to clarify what I mean by “no accumulation.” There is of course a sort of accumulation for all things at all times. You take a dose of some enteral medication, it leaches into your bloodstream from your gut, accumulating first in the serum. It then is distributed throughout the body and accumulates in other compartments (brain, liver, kidney, bone, whatever). Assuming linear pharmacokinetics, there’s some rate that the drug goes in to and out of each of these compartments.
If you keep taking the drug and the influx rate (from the serum into a compartment) is higher than the efflux rate (back to the serum from the compartment), the steady state in the compartment will be higher than the serum at steady state. In some sense, this could be called “accumulation.” But in another sense, if both these rates are fast, your accumulation is transient and quickly relaxes to zero if you clear the serum compartment of drug (which we know happens in normal individuals in the case of lithium). Although the concentration in the third compartment is indeed higher than in the serum, if you stop taking the drug, it will wash out (first from the serum then, more slowly, from the accumulating compartment).
SMTM: Thanks, this clarification is helpful. To make sure we understand, “accumulation” to you means that a contaminant goes to a part of the body, stays there, and basically never leaves. But you’re open to “a sort of accumulation” where 50 units go into the brain every day and only 10 units are cleared, leading to a more-or-less perpetual increase in the levels. Is that right?
JPC: Yes. I would frame this in terms of rates, though. So 5 x brain concentration units go to the brain and 1 x brain concentration units go out of the brain per unit time, such that you get a steady state concentration difference between the serum in the brain of in_rate / out_rate (in this case).
You guys seem mathy so I’ll add: for an arbitrary number of compartments this is just a first-order ODE. You can represent this situation as rate matrix K where element i, j represents the rate (1/time) that material flows from compartment i to j (or maybe j to i, I can never remember). Anyway this usually just boils down to something looking like an eigenvector problem to get the stationary distribution of things. (Obviously things get more complicated when you have pulsatile influx.)
The key question, though, is what effect does this high concentration in the accumulating compartment have on the actual physiology? If we have slowly-resolving, high concentration in the brain, then I think we could call this clinical (ie neuropharmacologically significant) accumulation. However, I think the case in the brain is that you have higher-than-serum concentrations, but that these concentrations quickly resolve after cessation of lithium therapy. My reasoning for this is that lithium pharmacokinetics are classically well-modeled with two- and three-compartment models, which mostly have pretty fast kinetics (rate parameters with half lives in the hours range).
SMTM: This is interesting because our sense is sort of the opposite! Specifically, our understanding is that most people who go off clinical doses of lithium do not lose much weight and tend to keep most of the weight they gained as a side effect (correct us if we’re wrong, we haven’t seen great documentation of this).
This seems at least suggestive that relatively high levels of lithium persist in the brain for a long time. On the other hand, clinical doses are really, really huge compared to trace doses, so maybe there is just so much in the brain compartment that it sometimes takes decades to clear. Ok we may not actually disagree, but it seemed like an interesting minor point of departure that might be worth considering.
JPC: I don’t know about this! I agree that slower (months to years) kinetics of lithium in the brain could explain this. An alternative (relatively parsimonious) explanation would be that, as Guyenet proposes, there simply is no mechanism for shedding excess adiposity. So if you gain weight as the result of any circumstance, if it stays on long enough for the lipostat to habituate to it, you just have a new, higher adiposity setpoint and have great difficulty eliminating that weight. That is, not being able to get the weight off after lithium-related weight gain might just be normal physiology.
The idea that clinical doses are just huge is sort of interesting. Normally, we think of the movement of ions in these kinetics models as having first-order kinetics (i.e. flux is proportional to concentration), but if you have truly shitboats of lithium in the brain, you could imagine that efflux might saturate (i.e. there are only so many transporters for the lithium to get out, since I imagine the cell membrane itself is impenetrable to Li+). This could be interesting. Not sure how you’d investigate it though. Probably patch-clamp type studies in ex vivo neurons? These are unfortunately expensive and extremely technical.
Amidsen
JPC: I see Amdisen et al. 1974 describes a fatal dose of lithium, which is very different pharmacokinetically from therapeutic doses. Above about 2.0 mmol/L (~2x therapeutic levels), lithium kinetics become nonlinear—that is, the pharmacokinetics are no longer fixed and the drug begins to influence its own clearance. In the case of lithium, high doses of lithium reduce clearance, leading to a vicious cycle of toxicity. This is a big deal clinically, often leading to the need for emergent hemodialysis.
So this is consistent with the papers I mentioned earlier (Ehrlich et al, Galliot et al) in the sense that cannot really conflict because they are reporting on two very different pharmacokinetic regimes.
You can’t directly compare the lithium kinetics in this patient to those in healthy people. You can see in figure 1 that the patient’s “urea” (I assume what we’d call BUN today?) explodes, which is a result of renal failure. It sounds like the patient wasn’t making any urine, i.e. has zero lithium clearance.
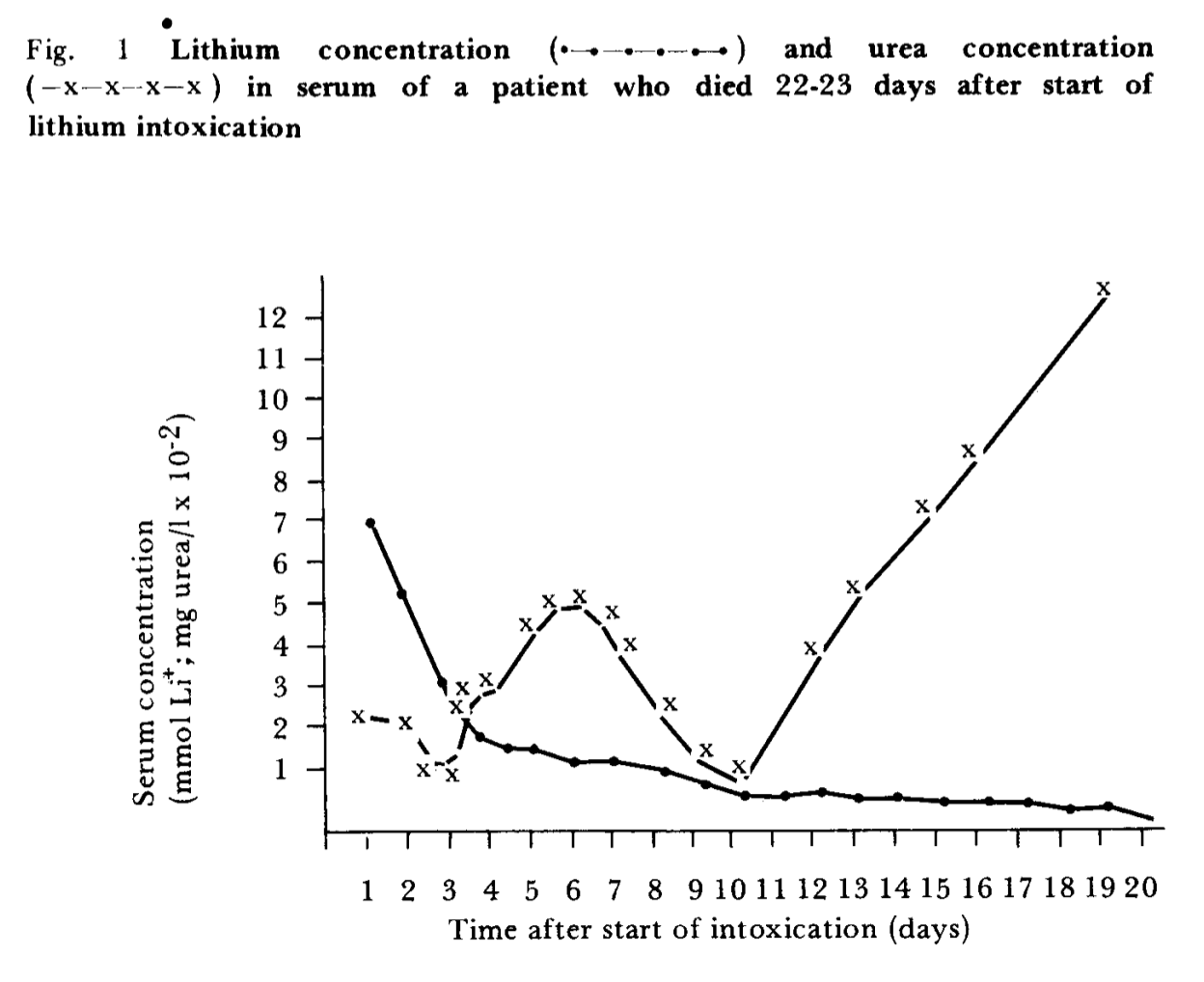
SMTM: True, it’s hard to tell. But FWIW lithium also seems to be cleared through other sources like sweat, so even renal failure doesn’t mean zero lithium clearance, just severely reduced. (Though not sure the percent. 50% through urine? 80%? 99%?)
JPC: Yes this is true, of course. My intuition would be that it’s closer to 99% or even like 99.9%. The kidney’s “function” (I guess you have to be a bit careful not to anthropomorphize/be teleological about the kidney here, but you know what I mean) is to eliminate stuff from the blood via urine, which it does very well, whereas sweat and other excreta have other functions.
Let’s assume for a second that lithium and sodium are the same and that the body doesn’t distinguish (obviously false; all models are wrong but some are useful) and let’s do some math.
In the ICU we routinely track “ins and outs” very carefully. Generally normal urine output is 0.5 – 1.5 mL/kg body weight/hr. In a 70 kg adult call it >800 mL/day. But because we also know how much fluid is going in, we know how much we lose to evaporation (sweat, spitting, coughing up gunk, etc), which we call “insensible losses.” This is usually 40-800 mL/day.
A normal sweat chloride (which we use to check for cystic fibrosis) is <29 mM. Because sweat doesn’t have a static charge, we know there’s some positive counterion. Let’s assume it’s all sodium. So call it 30 mM NaCl, and calculate 800 mL x 30 mM = 24 mmol NaCl and 40 mL x 30 mM = 1.2 mmol. These are collected using (I think) topical pilocarpine to stimulate sweat production, so this would be an upper bound probably. It’s pretty close to what they find here which is in athletes during training (full disclosure I didn’t read the whole thing), which seems like it would be similar to the pilocarpine case (i.e. unlikely to be sustained throughout the day).
We also measure 24-hour sodium elimination when investigating disorders of the kidney. A first-reasonabe-google-hit normal range is 40-220 mmol Na/24 hours. (Of course, this is usually done when fluid-restricting the patient, so this would be on the low end of normal. If you go to Shake Shack and eat a giant salty burger your urine urea and Na are going to skyrocket. If you’re in a desert, your urine will be WAY concentrated, but maybe lower volume. It’s hard to generalize so this is at best a Fermi estimation type of deal.)
Anyhow, we’re looking at somewhere between 2x and 250x more sodium eliminated in the urine. Again my guess is that we’d be closer to the 250x number and not the 2x number for some of the reasons I mention above. Also I worry you can’t just multiply insensible losses * sweat [Na] because as water evaporates it gets drawn out of the body as free water to re-hydrate the Na, or something.
In writing this up, I also found this paper which also does some interesting quantification of sweat electrolytes (again we get a mean sweat [Na] of 37 and [Cl] of 34), but in some of the later plots (Figure 2) we can see that [Na] and [Cl] go way low and that the average seems to be being pulled up by a long tail of high sweat electrolytes.
So not sure what to take away from that but I thought I’d share my work anyway. 🙂
Bone
JPC: In the case of bone, however, there might be something here! You could imagine the bone being a large but slowly-exchanging depot of lithium. I’d be interested to see if anyone has measured bone lithium levels in folks who were, say, on chronic therapeutic lithium. I’m not aware of anything like that.
SMTM: It seems to fit Amdisen et al. 1974. That case study is of a woman who was on clinical levels of lithium for three years, and had relatively high concentrations in her bones. Like you say, a fatal dose of lithium is very different pharmacokinetically from therapeutic doses, but the rate at which lithium deposits in bone is presumably (?) much slower than for other tissues, so this may be a reasonable estimate of how much had made it into her bones from three years of clinical treatment. Sample size of one, etc., but like you say there doesn’t seem to be any other data on lithium in bones.
JPC: I think it’s hard to say for sure if high concentration in her bones is due to the chronic therapy or the overdose. However, they note higher (0.77 vs 0.59 mmol/kg) in dense bone (iliac crest) than in spongey bone (vertebral body; there’s a better name than spongey… maybe cumulus? I don’t remember.). That’s interesting because it suggests to me (assuming that the error in the measurement is << 0.77-0.59) there is more concentrating effect in mineralized bone than all the cellular components (osteoclasts, osteoblasts, hematopoietic cells etc).
Anyway it’s suggestive that maybe there is deposition in bone. I wouldn’t hang my hat on it, but it is definitely consistent with it. I also agree that bone mineralization/incorporation seems like it ought to be on a longer timescale than cellular transport, so that is consistent as well. Obviously n=1, etc etc, but it’s kind of cute.
SMTM: Maybe we should see if we could do a study, there must be someone out there with a… skeleton bank? What do you call that?
JPC: A cadaver lab? I think most medical schools have them (ours does). In an academic medical setting, I would just get an IRB to collect bone samples from all the cadavers or maybe everyone who gets an autopsy that’s sufficiently extensive to make it easy to collect some bone. This would be a convenience sample, of course, but it would be interesting. Correlate age, zip code, renal function if known?
Because the patient is dead, there’s no risk of harm, and because they’re already doing the autopsy/dissection/whatever it should be relatively straightforward to collect in most cases (I mean, they remove organs and stuff to weigh and examine them so grabbing a bit of bone is easy). Unfortunately all these people got sick and died so you have a little bit of a problem there. For example, if someone had cancer and was cachectic, what can you learn from that? Idk.
In vivo bone biopsies are also a relatively common procedure done by interventional radiology under CT guidance (it’s SUPER COOL). You also have the problem that people are getting their biopsies for a reason, and usually the reason boils down to “we think that this bone looks weird,” so your samples would be almost by definition abnormal.
SMTM: Great! Maybe we can find someone with a cadaver lab and see if we can make it happen. This is a very cool idea.
Control Systems
SMTM: Earlier you mentioned the idea that the body’s set point can only be raised, but it seems really unlikely to us that there’s no mechanism for shedding excess adiposity.
JPC: Hmm. You guys are definitely better read on this subject than I am, but do I fear I have oversimplified the Guyenet hypothesis somewhat. My recollection is that it is more that there’s no driving force for the lipostat setpoint to return to a healthy level if it has habituated to a higher level of adiposity.
I like the analogy to iron. (I don’t think that Guyenet makes this connection, but I read The Hungry Brain years ago so I’m not sure.) It turns out that the body has no way of directly eliminating iron, so when iron levels get high, the body just turns off the “get more iron” system. Eventually, iron slowly makes its way out of the body because bleeding, entropy, etc etc and the iron-absorption system clicks back on. (This is relevant because patients who receive frequent transfusions, such as those with sickle cell, get iron overload due to their inability to eliminate the extra iron.)
I guess, by analogy, it would be that the mechanism for shedding adiposity would be “turn off the big hunger cues.” It’s not no mechanism, it’s just a crappy, passive, poorly-optimized mechanism. (Presumably because, like how nobody got transfusions prior to the 20th century, there was never an unending excess of trivially-accessible and highly palatable food in our evolutionary history.)
SMTM: Well, overfeeding studies raise people’s weights temporarily but they quickly go back to where they were before. Anecdotally, a lot of people who visit lean countries lose decent amounts of weight in just a few weeks. And occasionally people drop a couple hundred pounds for no apparent reason (if the contamination hypothesis is correct, this probably happens in rare cases where a person serendipitously eliminates most of their contamination load all at once). And people do have outlets like fidgeting that seem to be a mechanism beyond just “turn off the big hunger cues.” All this seems to suggest that weight is controlled in both directions.
JPC: Proponents of the above hypothesis would explain this by saying that the lipostat doesn’t have time to habituate to the new setpoint during the timescale of an overfeeding study, and so they lose the weight by having their “acute hunger cues” turned off. Whereas as weight creeps up year after year, the lipostat slowly follows the weight up. You do bring up a good point about fidgeting, though.
My thought was that bolus-dosed lithium (in food or elsewhere) might serve the function of repeated overfeeding episodes, each one pushing the lipostat up some small amount, leading to overall slow weight gain.
I think combining the idea that the brain concentrates lithium with an “up only” lipostat might give you this effect? If we say 1) lithium probably concentrates first in areas controlling hunger and thirst, leading to an effect on this at lower-than-theraputic serum concentrations, you might see weeks of weight-gain effect from a bolus 2) that we know that weight gain can occur on this timescale and then not revert (see the observation, which I read about in Guyenet, that most weight is gained between thanksgiving and NYE). What do you think?
SMTM: To get a little more into the weeds on this (because you may find it interesting), William Powers says in some of his writing (can’t recall where) that control systems built using neurons will have separate systems for “push up” and “push down” control. If he’s right, then there are separate “up lipostats” and “down lipostats”, and presumably they function or fail largely separately. This suggests that a contaminant that breaks one probably doesn’t break the other, and also suggests that the obesity epidemic would probably be the result of two or more contaminants.
JPC: Yes! Super interesting. There are lots of places in the brain where this kind of push-pull system is used. I remember very clearly a neuroscience professor saying, while aggressively waving his hands, that “engineers love this kind of thing and that’s probably why the brain does it too.” I wonder if he was thinking of Powers’ work when he said that.
SMTM: Let’s say that contaminant A raises the set point of the “down lipostat”, and contaminant B raises the set point of the “up lipostat”. Someone exposed to just A doesn’t necessarily get fatter, but they can drift up to the new set point if they overeat. At the same time, with exercise and calorie restriction, there’s nothing keeping them from pushing their weight down again.
Someone exposed to both A and B does necessarily get fatter, because they are being pushed up, and they have to fight the up lipostat to lose any weight, which is close to impossible. (This might explain why calorie restriction seems to work as a diet for some people but doesn’t work generally.)
Someone exposed to just B, or who has a paradoxical reaction to A, sees their up and down lipostats get in a fight, which looks like cycles of binging and purging and intense stress. This might possibly present as bulimia.
There isn’t enough evidence to tell to this level of detail, but a plausible read based on this theoretical perspective is that we might see something like, lithium raises the set point of the down lipostat and PFAS raise the set point of the up lipostat, and you only get really obese if you get exposed to high doses of both.
JPC: Very interesting! It’s definitely appealing on a theoretical level. (See: your recent post on beauty in science.) I just don’t know anything about the state of the evidence in the systems neuroscience of obesity to say if it’s consistent or inconsistent with the data. (Same is of course true of the lipostat-creep hypothesis above.)
I’m not sure about why you think the two systems would function separately? Certainly, for us to see a change, there would have to be a failure of one or the other population preferentially but I’m not sure why this would be less common than one effect or the other. They’d be likely anatomical neighbors, and perhaps even developmentally related. I guess it would all depend on the actual physiology. I’m thinking, for instance, of how the eye creates center-surround receptive fields using the same photoreceptors in combination with some (I think) inhibitory interneurons (neural NOT gates). The same photoreceptor, hooked up a different way, acts to activate or inhibit different retinal ganglion cells (the cells that make up the optic nerve… I think. It’s been a while.). Another example might be the basal ganglia, which (allegedly) functions to select between different actions, but mostly our drugs act to “do more actions” by being pro-dopaminergic (for instance to treat Parkinsons) or “do fewer actions” by being antidopaminergic (as in antipsychotics like haloperidol).
SMTM: Yeah good points and good question! We have reasons to believe that these systems (and other paired systems) do function more or less separately, but it might be too long to get into here. Long story short we think they are computationally separate but probably share a lot of underlying hardware.
Dynamics
SMTM: What do you think of a model based on peak lithium exposure? Our concern is that most sources of exposure are going to be lognormally distributed. Most of the time you get small doses, but very rarely you get a really really large dose. Most food contains no lithium grease, but every so often some grease gets on your hamburger during transport and you eat a big glob of it by accident.
Or even more concerning: you live downriver from a coal power plant, and you get your drinking water from the river. Most of the time the river contains only 10-20 ppb Li+, nothing all that impressive. But every few months they dump a new load of coal ash in the ash pond, which leaches lithium into the river, and for the next couple of days you’re drinking 10,000 ppb of lithium in every glass. This leads to a huge influx, and your compartments are filled with lithium.
This will deplete over time as your drinking water goes back to 10 ppb, but if it happens frequently enough, influx will be net greater than efflux over the long term and the general lithium levels in your compartments will go up and up. But anyone who comes to town to test your drinking water or your serum will find that levels in both are pretty low, unless they happen to show up on one of the very rare peak exposure days. So unless you did exhaustive testing or happened to be there on the right day, everything would look normal.
JPC: I totally vibe with the prediction that intake would be lognormally distributed. From a classic pharmacokinetic perspective, I would expect lognormally-distributed lithium boluses to actually be buffered by the fact that renal clearance eliminates lithium in proportion to its serum concentration–that is, it gets faster as lithium concentrations go up.
But I’m a big believer that you should shut up and calculate so I coded up a three compartment model (gut -> serum <-> tissue), made up some parameters* that seemed reasonable and gave the qualitative behavior I expected). Then either gave the model either 300 mg lithium carbonate three times a day (a low-ish dose of the the preparation given clinically), or three-times-a-day doses drawn from a lognormal distribution with two parameter sets (µ=1.5 and σ=1.5 or σ=2.5; this corresponds to a median dose of about 4.4 mg lithium carbonate in both cases, since the long tail doesn’t influence the median very much).
* k_gut->serum = 0.01 per minute
* k_serum->brain = 0.01 per minute
* k_brain->serum = 0.0025 per minute
* k_serum->urine = 0.001 per minute
* V_d,serum = 16 L
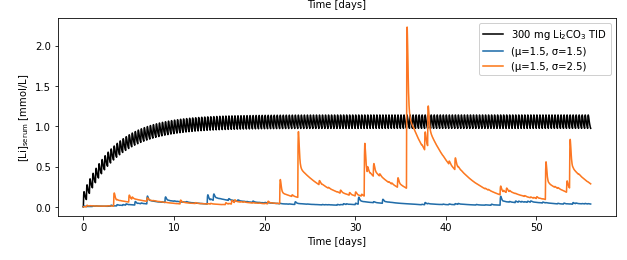
In my opinion, this gives us the following hypothesis: lognormally distributed doses of lithium with sufficient variability should create transient excursions of serum lithium into the therapeutic range.
Because this model includes that slow third compartment, we can also ask what the amount of lithium in that compartment is:
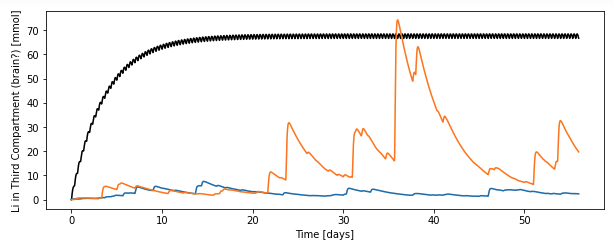
My interpretation of this is that the third compartment smooths the very spiky nature of the serum levels and, in that third compartment, you get nearly therapeutic levels of lithium in the third compartment for whole weeks (days ~35-40) after these spikes, especially if you get two spikes back to back. (Which it seems to me would be likely if you have, like, a coal ash spill or it’s wolfberry season or whatever.)
There clearly are a ton of limitations here: the parameters are made up by me, real kinetics are more like two slow compartments (this has one), lithium carbonate is a delayed preparation that almost certainly has different kinetics from food-based lithium, and I have no idea how realistic my lognormal parameters are, to name a few. However, I think the general principle holds: the slow compartment “smooths” the spikes, and so doing seems to be able to sustain highish [Li] even when the kidney is clearing it by feasting when Li is plentiful and retaining it during famine periods.
I’m not sure if this supports your hypothesis or not (do you need sustained brain [Li] above some threshold to get weight gain? I don’t think anyone knows…) but I thought the kinetics were interesting and best discussed with actual numbers and pictures than words. What do you guys think? Is this what you expected?
SMTM: Yes! Obviously the specifics of the dynamics matter a lot, but this seems to be a pretty clear demonstration of what we expected — that it’s theoretically possible to get therapeutic levels in the second compartment (serum) and sometimes in the third compartment (brain?), even if the median dose is much much lower than a therapeutic dose.
And because of the lognormal distribution, most samples of food or serum would have low levels of lithium — you would have to do a pretty exhaustive search to have a good chance of finding any of the spikes. So if something like this is what’s happening, it would make sense that no one has noticed.
It would be interesting to make a version of this model that also includes low-level constant exposure from drinking water (closer to 0.1 mg per day) and looks at dynamics over multiple years, getting an impression of what lifetime accumulation might look like, but that sounds like a project for another time.
Thyroid
JPC: Another thought is that thyroid concentrations may also matter. If lithium induces a slightly hypothyroid effect, people will gain weight that way too, since common (even classic) symptoms of hypothyroidism are weight gain and decreased activity. (It also proposes an immediate hypothesis [look at T3 vs TSH] and intervention [give people just a whiff of levothyroxine and see if it helps].) There’s also some thought that lithium maybe impacts thirst (full disclosure have not read this article except the abstract)?
SMTM: Also a good note, and yes, we do see signs of thyroid concentration. Some sort of thyroid sample would also be less invasive than a brain sample, right?
JPC: Yes. We routinely biopsy thyroid under ultrasound guidance for the evaluation of thyroid nodules (i.e. malignant vs benign). These biopsies might be a source of tissue you could test for lithium, but I’m not sure. The pathologists may need all the tissue they get for the diagnosis, they may not. Doing it on healthy people might be hard because it’s expensive (you need a well-trained operator) and more importantly it’s not a risk free procedure: the thyroid is highly vascular and if you goof you can hit a blood vessel and “brisk bleeding into the neck” is a pretty bad problem (if rare).
That said, it is definitely less invasive than a brain biopsy, and actually safer than the very low bar of “less invasive than a brain biopsy” implies.
Clinical
SMTM: Do you have clinical experience with lithium?
JPC: Minimal but non-zero. I had a couple of patients on lithium during my psychiatry rotation and I think one case of lithium toxicity on my toxicology rotation. I do know a lot of doctors, though, so I could ask around if they’re simple questions.
SMTM: Great! So, trace doses might be the whole story, but we’re also concerned about possible lithium accumulation in food (like we saw in the wolfberries in the Gila River Valley). We wonder if people are getting subclinical or even clinical doses from their food. We do plan to test for lithium in food, but it also occurred to us that a sign of this might be cases of undiagnosed lithium toxicity.
Let’s make up some rough numbers for example. Let’s say that a clinical dose is 600,000 µg and lithium toxicity happens at 800,000 µg. Let’s also say that corn is the only major crop that concentrates lithium, and that corn products can contain up to 200,000 µg, though most contain less. Most of the time you eat fewer than four of these products a day and get a subclinical dose of something like 50,000 – 300,000 µg. But one day you eat five corn products that all happen to be high in lithium, and you suddenly get 1,000,000 µg. You’ve just had an overdose. If common foods concentrate lithium to a high enough level, this should happen, at least on occasion.
If someone presents at the ER with vomiting, dizziness, and confusion, how many docs are going to suspect lithium toxicity, especially if the person isn’t on prescription lithium for bipolar? Same for tremor, ataxia, nystagmus, etc. We assume (?) no one is routinely checking the lithium blood levels of these patients for lithium, that no one would think to order this blood test. Even if they did, there’s a pretty narrow time window for blood levels detecting this spike, as far as we understand.
So our question is something like, if normal people are occasionally presenting with lithium toxicity, would the medical system even notice? Or would these cases be misdiagnosed as heavy metal exposure / dementia / ischemic stroke / etc.? If so, is there any way we can follow up with this? Ask some ER docs to start ordering lithium tests in any mystery cases they see? Curious to know what you think, if this seems at all plausible or useful.
JPC: I have a close friend who is an ED doc! She and I talked about it and here’s our vibe:
With a presentation as nonspecific as vomiting, dizziness, and confusion, my impression is that most ED docs would be unlikely to check a lithium level, especially if the patient is well enough to say convincingly “no I didn’t take any pills and no I don’t take lithium.” At some point, you might send off a lithium level as a hail-Mary, but there are so many things that cause this that a very plausible story would be: patient comes to ED with nausea/vomiting, dizziness, and altered mental status. The ED gives maybe fluids, checks some basic labs, does an initial workup, and doesn’t find anything. Admits the patient. The next day the admitting team does some more stuff, checks some other things, and comes up empty. The patient gets better after maybe 24-48h, nobody ever thinks to check a lithium level, and since the patient is feeling better they’re discharged without ever knowing why.
Another version would go: patient is super sick, maybe their vomiting and diarrhea get them super dehydrated and give them an AKI (basically temporary kidney failure). People think “wow maybe it’s really bad gastritis or some kind of primary GI problem or something?” The patient is admitted to the ICU with some kind of gross electrolyte imbalance because they’re in kidney failure and they pooped out all their potassium, someone decides they need hemodialysis, and this clears the lithium. Again the patient gets better, and everyone is none the wiser.
Tremor, ataxia, nystagmus, etc. are more focal signs and even if someone doesn’t have a history of lithium use, and in this case our impression is that people would be more likely to check a lithium level. We also think it wouldn’t always happen. Even in classic presentations of lithium toxicity, sometimes people miss the diagnosis. (Emergency medicine is hard; people aren’t like routers where they blink the link light red when the motherboard is fried or power light goes orange if the AC is under voltage. Things are often vague and complicated and mysterious.)
Something you’d have to explain is how this isn’t happening CONSTANTLY to people with really borderline kidney function. Perhaps one explanation might be that acute lithium intoxication (i.e. not against a background of existing lithium therapy) generally presents late with the neuro stuff (or so I hear).
We think that this is plausible if it is relatively uncommon or almost always pretty mild. If we were having an epidemic of this kind of thing (like on the scale of the obesity epidemic) I think it would be weird that nobody has noticed. Unless of course it’s a pretty mild, self-resolving thing. Then, who knows! AFAIK still nobody really knows why sideaches happen—figuring it out just isn’t a priority.
On occasion, the medical-scientific community also has big misses. There’s an old line that “half of what you learn in medical school is false, you just don’t know which half.” We were convinced until 1982 that ulcers were caused by lifestyle and “too much acid”; turns out that’s completely wrong and actually it’s bacteria. I saw a paper recently that argued that pretty much all MS might be due to EBV infection (no idea if it’s any good).
I think you could theoretically “add on” a lithium level to anybody that’s getting a head CT with the indication being “altered mental status.” “Add on” just means that the lab will just take the blood they already have from the patient and run additional testing, if they have enough in the right kind of tube. The logic is that patients with new-onset, dramatic, and unexplained mental status changes often get head CTs to rule out a bleed or other intracranial badness, so a head CT ordered this way could be a sign that the ordering doc may be feeling stumped.
If you wanted to get fancy, you could try to come up with a lab signature of “nausea/vomiting/diarrhea of unclear origin” (maybe certain labs being ordered that look like a fishing expedition) and add on a lithium there as well.
SMTM: Good point, but, isn’t it possible that it IS happening constantly to people with really borderline kidney function? The symptoms of loss of kidney function have some overlap with the symptoms of lithium intoxication, maybe people with reduced kidney function really do have this happen to one degree or another whenever they draw the short straw on dietary lithium exposure for the day. Lots of people have mysterious ailments that lead to symptoms like nausea and dizziness, seemingly at random.
Or we could look at it from the other angle — lithium can cause kidney damage, kidney disease is (very roughly) correlated with obesity at the state level, and as far as we can tell, rates of kidney disease are going up, right? Is it possible that many cases interpreted as chronic kidney disease are “actually” chronic lithium intoxication?
JPC: I guess it’s definitely possible. The “canonical” explanation to this would be that diabetes (which is obviously linked to obesity) destroys your kidneys. But, if it’s all correlated together as a vicious cycle (lithium → obesity → CKD → lithium) that’s kind of appealing too. I bet a lot is known about the obesity-diabetes-kidney disease link though and my bet without looking into it would be that there’s some problem with that hypothesis.
My thought here was that if people with marginal/no kidney function are getting mild cases, I would expect people with normal kidney function to be basically immune. Or, if people with normal kidney function get mild cases, people with marginal kidneys should get raging cases. This is because serum levels of stuff are related to the inverse of clearance. The classic example is creatinine, which is filtered by the kidney and used as a (rough) proxy for renal function.
SMTM: This is super fascinating/helpful. For a long time now we’ve been looking for a “silver bullet” on the lithium hypothesis — something which, if the hypothesis is correct, should be possible and would bring us from “plausible” to “pretty likely” or even “that’s probably what’s going on”. For a long time we thought the only silver bullet would be actually curing obesity in a sample population by making sure they weren’t consuming any lithium, but that’s a pretty tall order for a variety of reasons, not least because (as we’ve been discussing) the kinetics remain unclear! But recently we’ve realized there might be other silver bullets. One would be finding high levels of lithium in food products, but there are a lot of different kinds of foods out there, and since the levels are probably lognormal distributed you might need an exhaustive search.
But now we think that finding people admitted to the ER with vague symptoms and high serum lithium, despite not taking it clinically, could be a silver bullet too. Even a single case study would be pretty compelling, and we could use any cases we found to try to narrow down which foods we should look at more closely. Or if we can’t find any of these cases, a study of lithium levels in thyroid or in bone could potentially be another silver bullet, especially if levels were correlated with BMI or something.
JPC: I’m always hesitant to describe any single experiment as a silver bullet, but I agree that even a single case report, under the right conditions, of high serum lithium in someone not taking lithium would be pretty suspicious. You’d have to rule out foul play and primary/secondary gain (i.e. lying) but it would definitely be interesting. As far as finding lithium in bone or thyroid (of someone not taking lithium), I’d want to see some kind of evidence that it’s doing something, but again it’d definitely be supportive.
SMTM: Absolutely. We also don’t really believe in definitive experiments. The goal at this stage is to look for places where there might be evidence that could promote this idea from “plausible” to “likely”.

TLDR based on my personal experience all of this makes the lithium hypothesis weaker for me, as I gained weight on lithium and them lost it, stabilizing at a much lower lipostat level.
I was on high doses of lithium for bipolar for about two years. During this time I gained weight, up to 185lb (BMI 26) over my normal adult set point of 155 lb (BMI~21). I got into macro counting and weight lifting, and about 6 months later, stopped taking lithium. I lost a lot of weight in the next year, down to about 127lb (BMI 17), and now 3.5 years and one pregnancy later, I am stable at 140 lb (BMI 19), lower than my start point. This to me makes the idea of brain/bone lithium accumulation unconvincing. I lived in the same place during this five year period (Boston). My diet has however changed to become much less nutrient dense (e.g. cauli rice not real rice, avoiding caloric foods) so I stay leaner perhaps because despite a constant amount of hunger I have found a way to eat fewer calories and feel fuller (falling into the 10 lb “I can control it” range. But I think that can account for my lower than high school weight now, not necessarily my ability to loose the weight in the first place. Of course N=1 but it is incorrect to assume people just keep getting heavier after lithium.
Not a scientific paper but since we’re trying to thin out of the box I’d be curious on your thoughts on Stephanie Buttermore’s controversial “all in” diet. She was a bikini competitor who developed “extreme hunger” after chronic dieting (e.g. lipostat set high) and ate and gained weight until the extreme hunger was sated, and then slowly stabilized at a normal weight (lowered lipostat). I do not know where she lived during the experiment though. 🙂 https://youtu.be/0SNJjEIoGFs
LikeLike
Jesus Christ the ebv link — it looks solid to me. Did they just figure out MS?
LikeLike
So glad you are continuing to work on this super interesting and important obesogen hypothesis. I have definitely experienced the phenomenon of sudden effortless weight loss associated with a short vacation to a lean country! I am curious how likely you think that the obesogen is primarily in water vs. in food vs. other sources. I’m interested in trying to reduce lithium/PFA exposure but of course don’t want to waste my time on sources that are insignificant relative to total exposure. Also curious what filtration methods work against PFAs or lithium.
LikeLike
Of course! Yeah we would love to know the answers to those questions too but so far we just don’t know. As soon as we learn anything new we will put it up on the blog!
LikeLike
Anecdotal re: thyroid issues
I am obese. I have been overweight since grade school. I grew up in one of the cities you listed as top 10 obesity percentage. My family who live in that city are all obese or overweight. We all drink tap water. My family who did not ever live in that city are normal weight.
I have thyroid issues. They run in my family. I am technically subclinical hypothyroid as my T4/T3/TSH are just on the edge of normal range.
Eventually a doctor put me on low dose unithroid. I immediately saw improvement in my symptoms. Although I still struggle with hunger and satiety, it is less intense. And my energy levels and coldness have improved.
LikeLike