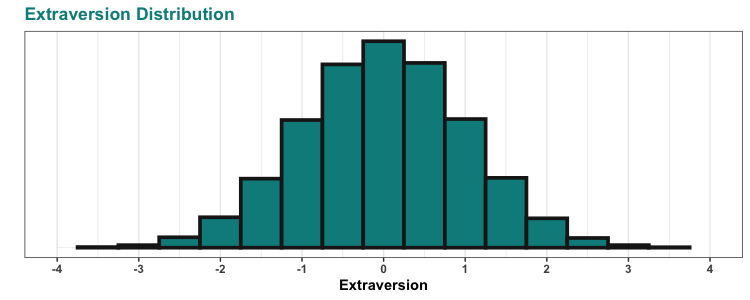
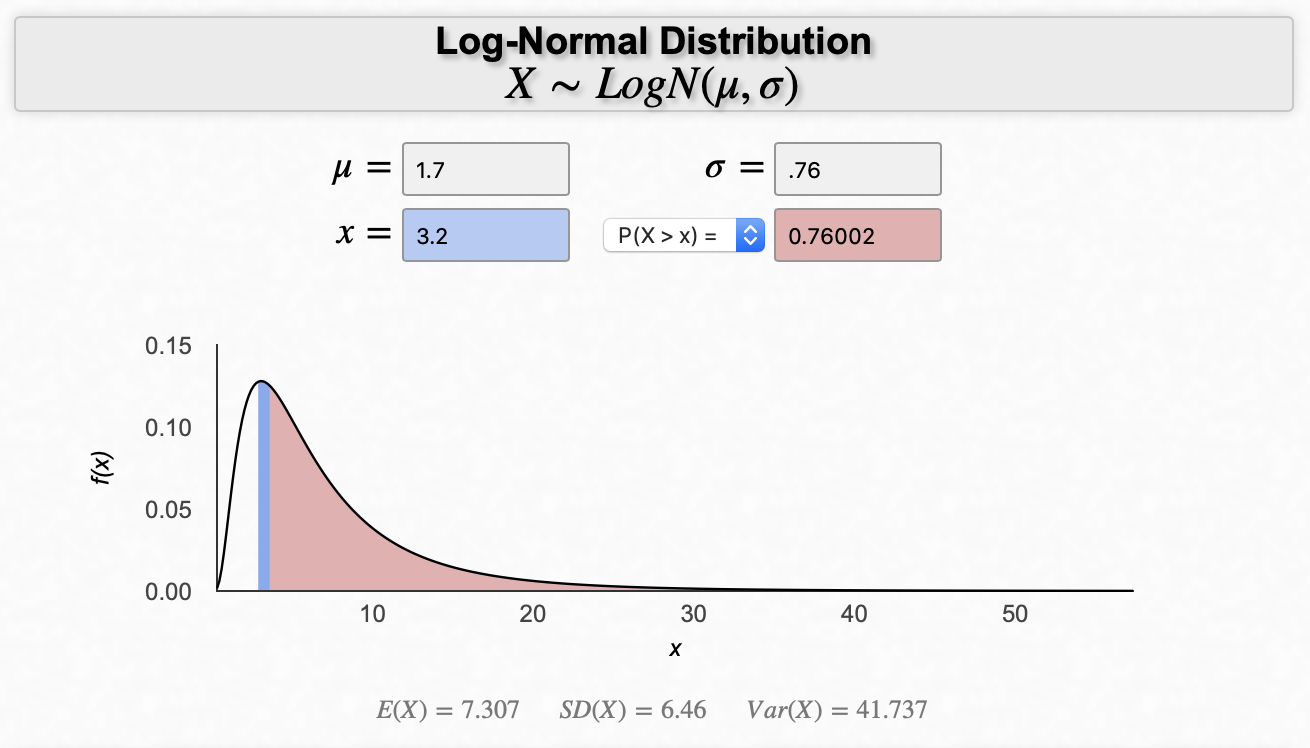
So, the CDC has this project called the National Health and Nutrition Examination Survey, or NHANES for short. They describe the project as:
…a program of studies designed to assess the health and nutritional status of adults and children in the United States. … [It] began in the early 1960s and has been conducted as a series of surveys focusing on different population groups or health topics. In 1999, the survey became a continuous program that has a changing focus on a variety of health and nutrition measurements to meet emerging needs. The survey examines a nationally representative sample of about 5,000 persons each year. These persons are located in counties across the country, 15 of which are visited each year.
Data from the NHANES is publicly available. It includes hundreds of health and nutrition measures for thousands of people, collected in multiple rounds of examination, across several decades.

But NHANES data is very hard to work with for the same reasons. Each two-year period of data (e.g. 1999-2000, 2001-2002, etc.) is split up into several different datasets, which have to be combined for analysis. Comparing measures across multiple years can be quite tricky, because formatting and variable names often change year-to-year, sometimes with no explanation. Variables are often added or removed, and it’s not always clear if a measure used in one year is the same as a similarly-named measure in another year. So while the dataset is extremely rich, its hugeness can make it difficult to work with.
To address these problems, we worked with a data scientist, Elizabeth, to combine the NHANES datasets from 1999 to 2018 into a single package. The dataset we ended up with is certainly imperfect — we didn’t include every measure, we still aren’t sure which variables are duplicates, etc. — but it’s a starting point.
Now, Elizabeth and the SMTM team are going through the dataset to see what it can tell us about the obesity epidemic, to see if there are any new mysteries we can uncover, and in particular to see what it can tell us about the contamination hypothesis of obesity. Analysis was performed only on data from adults (people 18 or over), and not including anyone marked as pregnant. Elizabeth is great, all mistakes are ours not hers, etc.
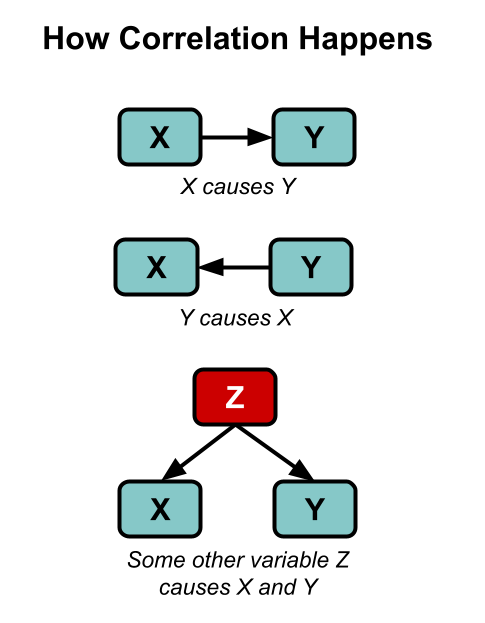
Here’s our standard disclaimer: all of the analysis you are about to see is correlational. As you well know, correlation does not imply causation — though as XKCD reminds us, “it does waggle its eyebrows suggestively and gesture furtively while mouthing ‘look over there’.” Correlation can still provide some evidence, and we can always follow up on anything we find in correlational data later on with more controlled research. We are fishing for weird surprises, like when you cast a net into the sea and pull up a bicycle.
A final reminder: about 75% of the modern variation in BMI is genetic, so any correlations of environmental variables with BMI will probably appear to be quite small. We should expect to see correlations like r = 0.10, not like r = 0.65. But that appearance of smallness is misleading, because we are looking for factors that can explain the 25% of the variance in BMI that isn’t genetic. If we find a factor that accounts for 5% of the total variance in BMI, it’s more like it explains 20% of the remaining mystery, the non-genetic variance (.05/.25 = 0.2), which is what we are really interested in.
We started with the simplest possible analyses. In this case it was as simple as: in the NHANES data, which variables have the biggest correlations with BMI?
The strongest correlations of all, of course, were correlations of BMI with other measures of obesity. Naturally BMI is correlated with things like height and weight, this is not a surprise. So let’s pass over these variables and move on to more interesting findings.
From here, two correlations stand out in particular:
Serum copper levels have the strongest correlation with BMI in the dataset, r = 0.240 (p < .001), and serum copper levels account for about 5.8% of the variance in BMI.
Serum gamma-tocopherol levels are a close second, correlated with BMI at r = 0.230 (p < .001), and serum gamma-tocopherol levels account for about 5.5% of the variance in BMI.
Both of these relationships stand out quite a bit from the herd. The next-strongest correlation with BMI in the NHANES is just r = -0.170 (we’ll look at some of these variables in future posts). This is still significant but it’s definitely a step down — serum copper and serum gamma-tocopherol are, for some reason, pretty clear outliers.
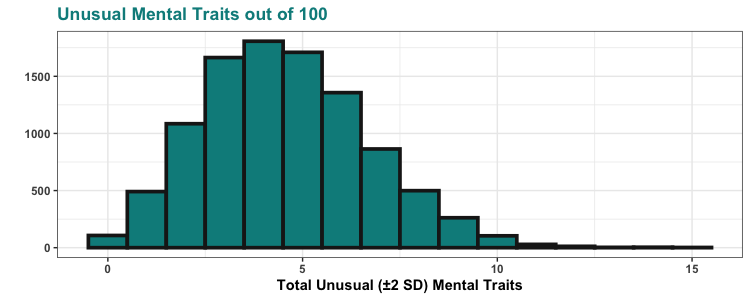
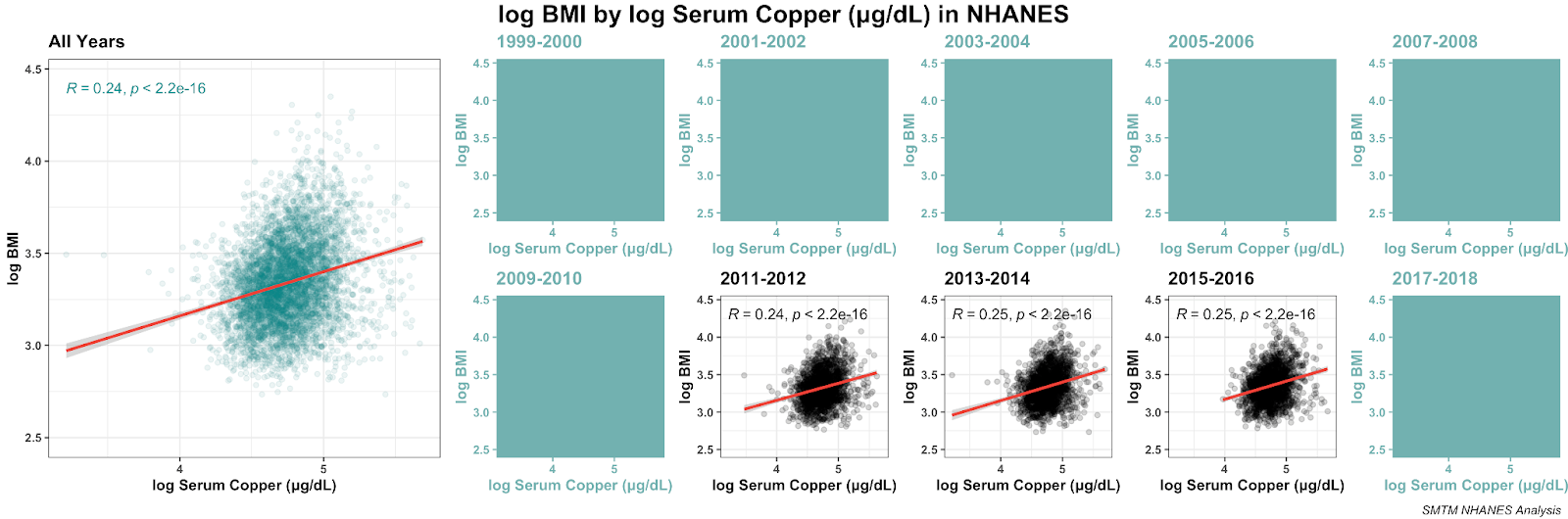
Here’s a plot of the 20 strongest correlations with BMI in the NHANES data that we looked at (not including other measures of obesity). There are plenty of variables correlated with BMI, but serum copper and serum gamma-tocopherol are the only correlations above .20 and really stand out when you plot them graphically. Here they are plotted with absolute value, so we can compare positive and negative correlations without worrying about the sign:
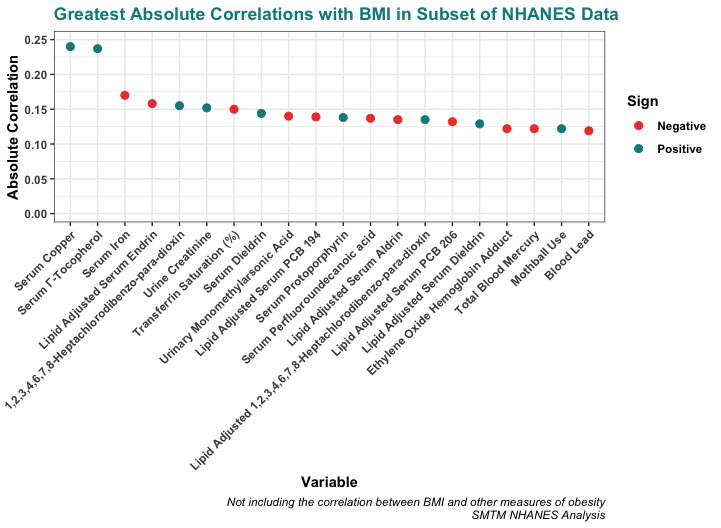
These correlations might seem small, but remember that about 75% of the variation in BMI is genetic. These small correlations are actually quite big in context. For example, the correlation between BMI and age (people generally get heavier as they get older) is only r = 0.072 — all the correlations pictured above are a bit larger than this.
This seems particularly interesting because while these two correlations are potentially compatible with a number of different theories, we don’t think there’s any theory of the obesity epidemic that would actually have predicted these correlations in advance.
Sadly we cannot compare copper and gamma-tocopherol directly since, as you will see in a minute, they cover different years of the dataset. But we can look at them individually, so let’s do that.
Copper
The NHANES only collected serum copper in three datasets: 2011-2012, 2013-2014, and 2015-2016. But these provide three mostly independent replications, and the same relationship is found all three times.
Here’s that relationship with both variables log-transformed. The log transformation makes the visualization clearer, but the correlation remains pretty much the same:
And here’s the distribution of serum copper levels in those three datasets:
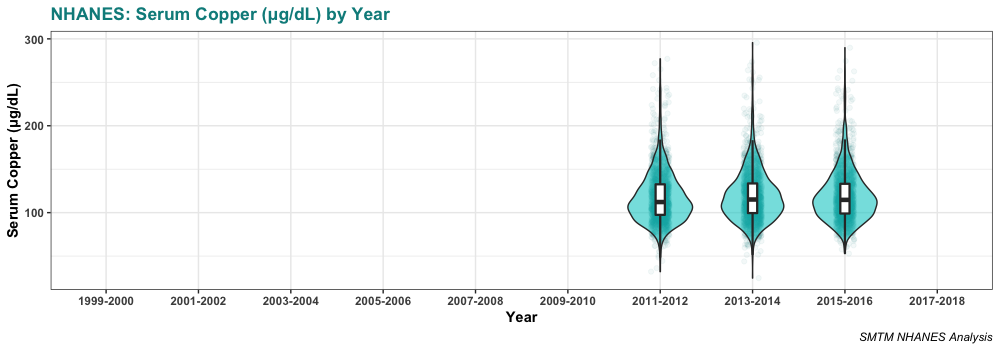
Serum copper levels are lowest in 2011-2012, the first year we have data for. They might be increasing a bit over time, but really there’s not enough years of data to tell.
Elizabeth did a different kind of visualization, where she took a look at the individual deciles for copper (she split people up into 10 groups by how much copper was in their serum) and found that the relationship is even more striking with this kind of analysis.
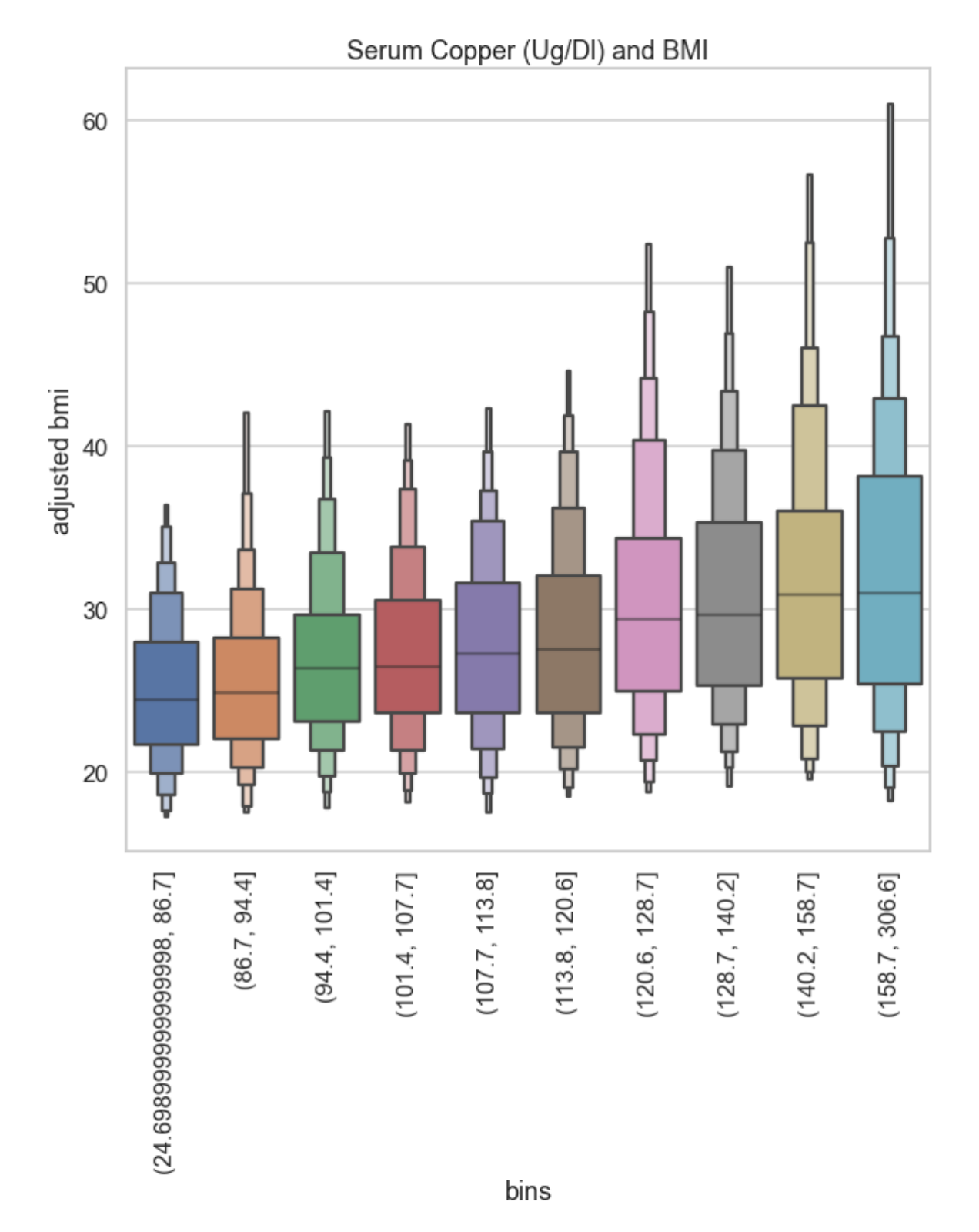
The people in the lowest two brackets for serum copper levels have median BMIs of 25, and around 70% of them have BMIs under 30.
In comparison, higher levels get you a higher median and a bigger spread — the top two brackets for serum copper levels have a median BMIs over 30. People in the top 20% for serum copper are on average obese. This is very weird.
Finally, if you put serum copper in a regression model with age, gender, ethnicity, and education, it remains significant and the coefficient suggests that serum copper levels account for about 3.9 points of BMI difference across the normal range of serum copper levels. The model as a whole explains about 11% of the variation in BMI in this dataset.
You might think that this correlation is driven by people with another condition that manifests as high serum copper, like kidney disease. But in fact, around 80% of people in this sample are within the normal range. If we look at just the people in the normal range (62-140 μg/dL), we get almost exactly the same correlation with BMI, r = 0.241. It seems like the BMI / serum copper relationship exists within the normal parameters and the general population.
Biology
Copper does a bunch of stuff in the human body, so looking at its biological role does not narrow things down much at all. Per Wikipedia:
Copper is incorporated into a variety of proteins and metalloenzymes which perform essential metabolic functions; the micronutrient is necessary for the proper growth, development, and maintenance of bone, connective tissue, brain, heart, and many other body organs. Similarly to some other divalent ions, copper strongly interacts with lipid membranes and is involved in the formation of red blood cells, the absorption and utilization of iron, the metabolism of cholesterol and glucose, and the synthesis and release of life-sustaining proteins and enzymes. These enzymes in turn produce cellular energy and regulate nerve transmission, blood clotting, and oxygen transport.
This is additionally weird to us because we’re talking about serum copper levels. As an essential trace element, serum copper should be under tight homeostatic control, and really should not be correlated with much of anything. Wikipedia mentions this too: “The human body has complex homeostatic mechanisms which attempt to ensure a constant supply of available copper, while eliminating excess copper whenever this occurs.”
One way to interpret this could be that serum copper is not directly related to BMI at all — it could be that there’s something else that makes you obese AND messes up the control system that should keep your serum copper levels in the right range.
This is supported by the fact that the correlation between BMI and dietary copper is negative and nearly zero (r = -0.035). (Though also bear in mind imperfect collecting methods — all the nutrition measures are based on dietary interviews, so the estimates of elements are probably not very accurate.)
We brought this finding to a doctor we know, hoping he could shed some light on the result, but he was also pretty mystified. We also brought it to a biochemist, and she said, “Woah!!” (She also pointed out that copper is kind of an unusual ion because it has a couple of different charge levels.)
Literature Review
With the help of these colleagues, we started looking through the literature, and it turns out there’s already a small body of research out there about serum copper and obesity. We’re not even the first researchers to notice this in the NHANES data — this team of Chinese researchers wrote about serum copper and obesity in the NHANES data in 2017. But most of these papers haven’t gotten much attention.
The oldest source we saw was this paper from 1997, by a Turkish team studying children in Turkey. They found that serum copper concentrations were significantly higher in obese children than in “healthy controls”. They also report a similar relationship for serum zinc. They don’t mention any previous work linking copper to obesity.
This paper from a Tunisian team in 2001 found higher levels of serum copper in a group of obese participants. While they don’t report a correlation, they do mention that people with higher BMIs tend to have higher serum copper levels.
This paper from 2003, by a team from Kuwait University, found that serum copper concentration was associated with BMI (r = 0.220, p < 0.001). That’s really similar to the correlation we see in the NHANES data, r = 0.240. They also report some other relationships between leptin (one of the hormones that regulates fat storage) and various measures, including the serum zinc/copper ratio. It does seem a bit like they’re just picking variables and searching for relationships at random, but it’s still very encouraging to see this weird relationship we found in the NHANES replicated in a Kuwaiti population from almost a decade earlier.
This paper from 2019, by a team at Johns Hopkins, found a “significant positive and element-specific correlation between copper and BMI after controlling for gender, age, and ethnicity. Serum copper also positively correlated with leptin, insulin, and the leptin/BMI ratio.” They actually find a stronger relationship than in the NHANES, r = 0.40.
Finally, this meta-analysis from 2019 looked at 21 previous publications and found that serum copper is generally related to obesity. They speculate that “elevated [copper] in circulation could contribute to oxidative stress disorder that reflects free radical concentrations and antioxidant imbalance” as well as some other mechanisms. That’s nice and all, but it doesn’t really explain why copper levels would be elevated in the first place.
(Of interest to previous discussions, they also found that “in stratified analysis by the detection methods, the results showed that the association of serum Cu and obesity was significantly detected by AAS including flame atomic absorption spectrometry (FAAS) … but not for studies detected by ICP-MS.”)

One thing that stands out here is that many of these papers mention zinc. Copper and zinc are antagonists, meaning they work against each other in the body, so if copper is somehow involved in the obesity epidemic, it would make sense if zinc were involved too.
However, we don’t see much evidence for it in the NHANES data. Serum zinc is not very correlated with serum copper or with BMI, though we noticed that if you put serum zinc and serum copper in a regression model predicting BMI, the interaction is significant (though only p = .004 with n > 5000). The idea that zinc is part of the picture remains interesting but tenuous.
So much for copper.
Γ-Tocopherol
Γ-Tocopherol (gamma-tocopherol) is a form of vitamin E. Vitamin E is not a single compound, but a family of eight different compounds, four tocopherols and four tocotrienols (and also a secret third form of vitamin E, the synthetic tocofersolan). Gamma is the third letter in the Greek alphabet, and gamma-tocopherol is the third tocopherol in the vitamin E family.
Along with serum copper levels, serum concentration of gamma-tocopherol stands out in the NHANES data for having an unusually strong correlation with BMI, r = 0.230, or r = 0.242 if both variables are log-transformed.
Unfortunately, the gamma-tocopherol data is a little confusing, because NHANES kept changing the names of the serum tocopherol measures and the datasets they were a part of. Just because something is in a public dataset doesn’t mean it’s trivial to organize and make sense of it. If anything, quite the opposite. Let’s quickly disentangle.
In 1999-2000, gamma-tocopherol is in the Cadmium, Lead, Mercury, Cotinine & Nutritional Biochemistries (LAB06) dataset. There’s also a variable just called “Vitamin E” which is probably alpha-tocopherol, but we don’t see anything that makes this explicit.
In 2001-2002, gamma-tocopherol is in the Vitamin A, Vitamin E & Carotenoids (L06VIT_B) dataset as “g-tocopherol”, and alpha-tocopherol is in this dataset explicitly as “Vitamin E”. Both alpha- and gamma-tocopherol are ALSO in the Vitamin A, Vitamin E, & Carotenoids, Second Exam (VIT_2_B) dataset, but in this dataset they are called “alpha-tocopherol” and “gamma-tocopherol”. These seem to be the same measures taken on a second day of testing, which is kind of neat, but you’d think they could keep the variable names consistent within a single year’s data.
In 2003-2004, alpha-, gamma-, and delta-tocopherol are in the Vitamin A, Vitamin E & Carotenoids (L45VIT_C) dataset as “a-tocopherol”, “g-tocopherol”, and “d-tocopherol”, respectively.
In 2005-2006, gamma-tocopherol is in the Vitamin A, Vitamin E & Carotenoids (VITAEC_D) dataset as “g-tocopherol”, and alpha-tocopherol is in the same dataset but they’re calling it “Vitamin E” again.
In 2007-2008, 2009-2010, 2011-2012, and 2015-2016, there don’t appear to be any serum measures of tocopherols (there are dietary measures, but we don’t have much confidence in those). Oops?
However, serum measures reappear in the 2017-2018 data. Both alpha- and gamma-tocopherol appear in Vitamin A, Vitamin E & Carotenoids (VITAEC_J) as “alpha-tocopherol” and “gamma-tocopherol”.
(The NHANES is very rich but, as you might have noticed, rather disorganized. If you 1. have the data / database chops and are interested in cleaning it up, 2. have medical / biochemical / nutritional / etc. expertise and are interested in organizing the measures across years, or 3. are interested in funding a project to clean up and organize this mess so other people can more easily do the sort of thing we’re doing here, let us know and we’ll put you in touch with one another.)
While the naming conventions across these years are very messy, we didn’t see anything to suggest that these variables were measured in any substantially different way in different years. The 1999-2000 dataset seems to be ambiguous, but the others all describe the analysis method as being “high performance liquid chromatography with photodiode array detection”. So we combined all the tocopherol measures across years into single variables that we will use from here on out.
Here’s the overall relationship between BMI and gamma-tocopherol as a scatterplot. As usual, we have log-transformed both variables for a cleaner visualization:
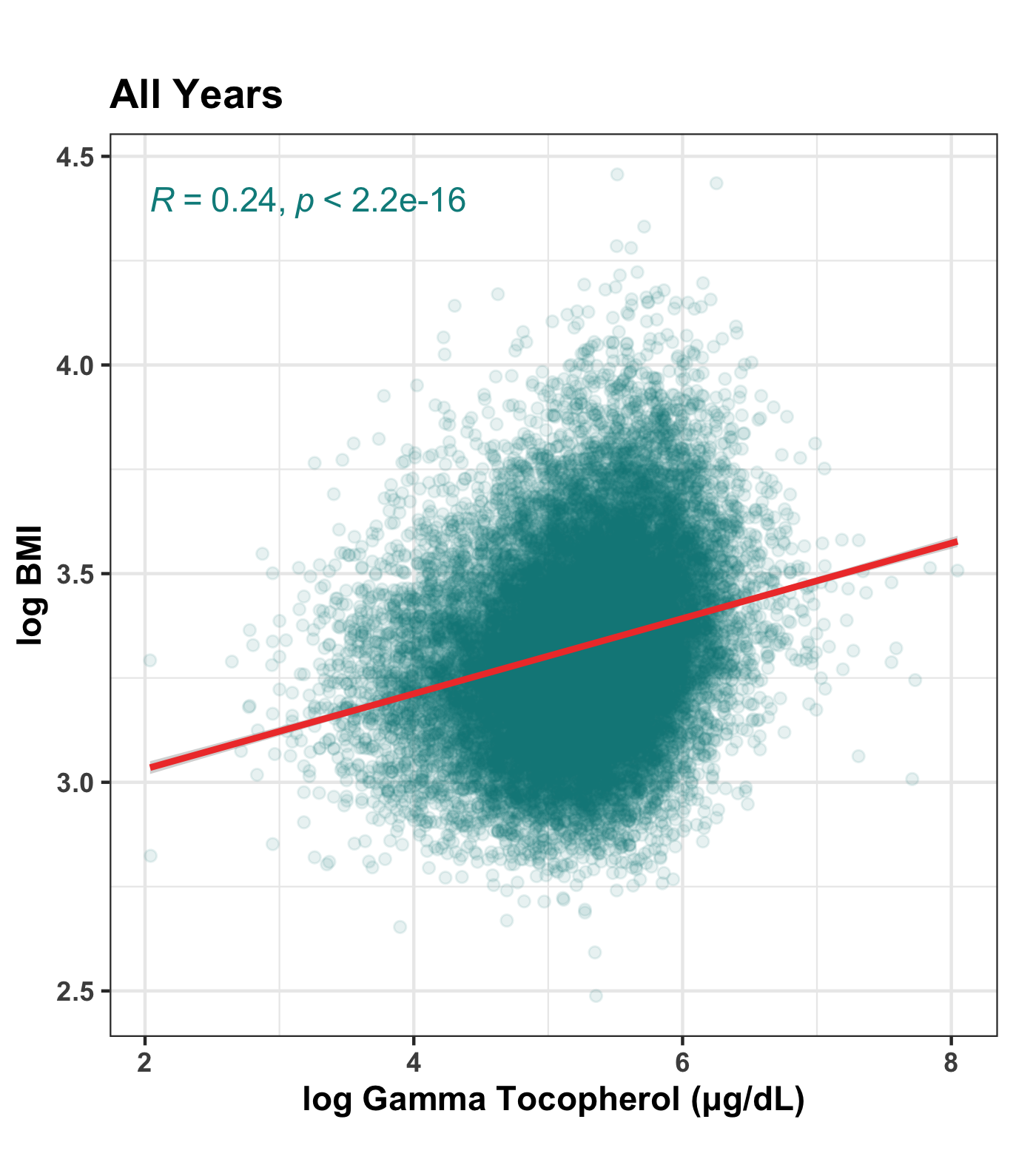
Here it is broken out by year:
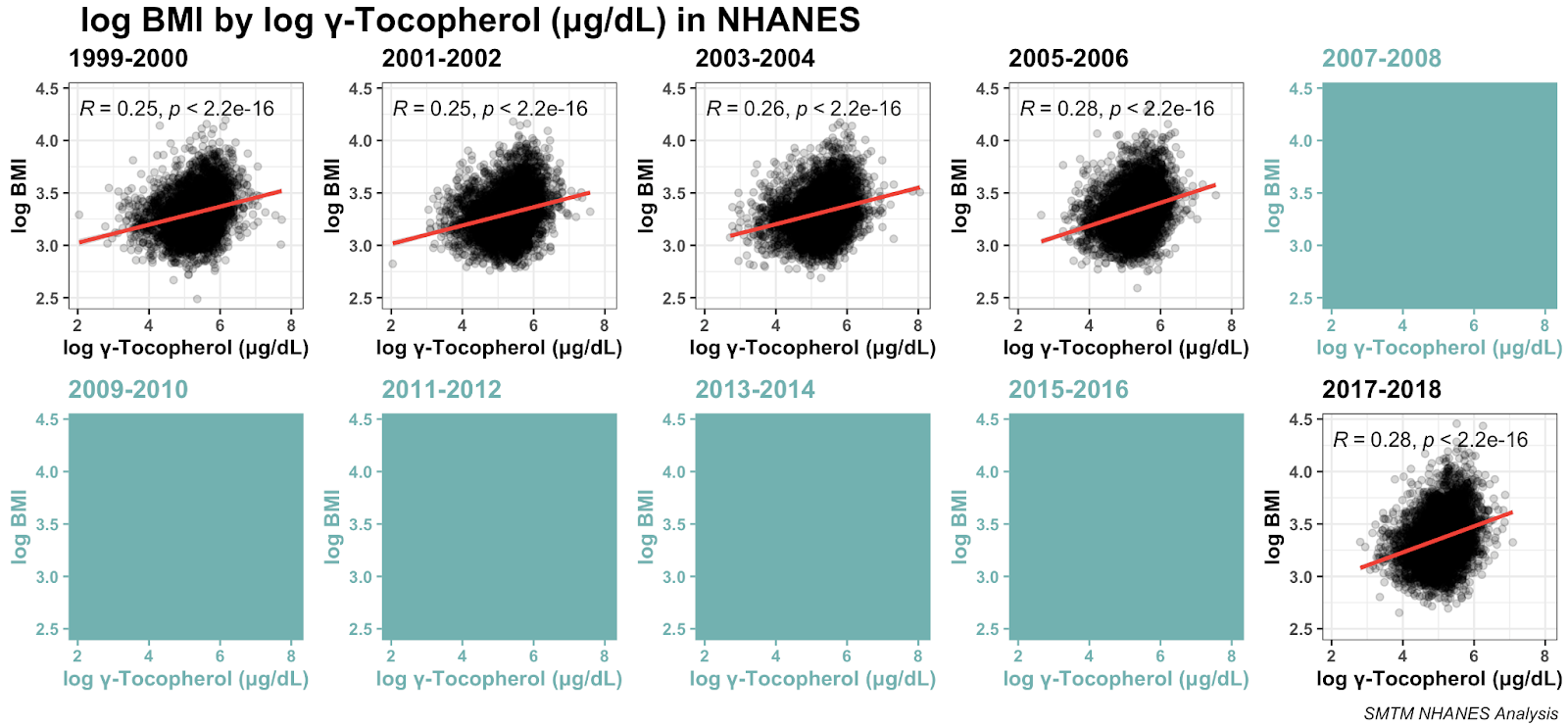
As we can see, the relationship seems to be very robust, replicating across five different datasets.
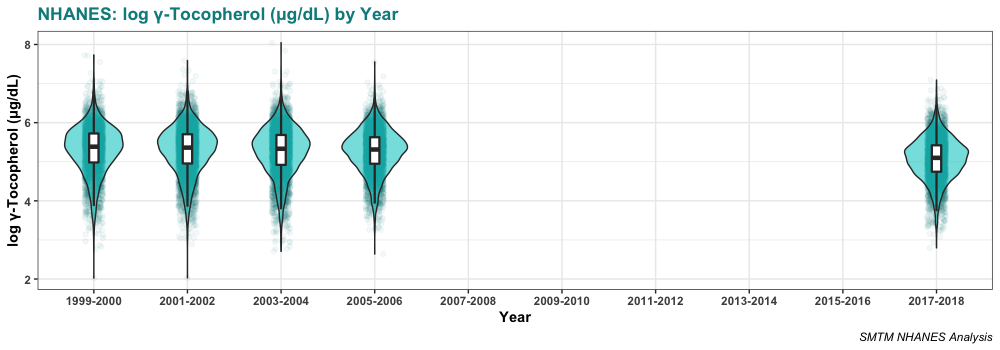
Serum levels of gamma-tocopherol seem to be declining slightly over time. The average was 240.0 µg/dL in 1999-2000 and is only 181.9 µg/dL in 2017-2018. This is certainly one point against the idea that gamma-tocopherol is causing obesity. If it were causing obesity, you would expect gamma-tocopherol levels to increase over time, as obesity has increased.
Here’s the distribution in those years:
Elizabeth used a more advanced technique and found the following:
A 2 variable decision tree for that year sometimes splits up on just Gamma Tocopherol and Iron (only time Iron shows up directly), which is an unexpected level of importance.
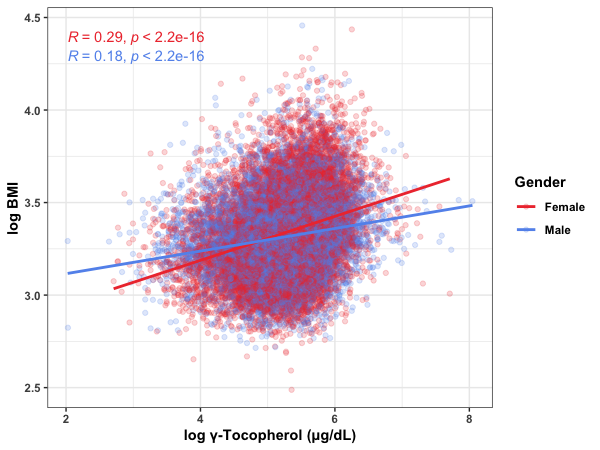
Less restricted decision trees put Gamma Tocopherol levels after a split on gender, with a 4 point difference in predicted BMI. Some of the literature suggests that the effect may be gender specific, so that’s interesting.
She’s right: the relationship is stronger for women than it is for men. The correlation between BMI and serum gamma-tocopherol is r = 0.18 for male participants and r = 0.29 for female participants. In a multiple regression, this interaction is clearly significant, p < 0.001.
And like Elizabeth mentions, this is alluded to in some of the existing literature. This paper looking at the British National Diet and Nutrition Survey also finds a relationship between serum gamma-tocopherol and BMI, and highlights it for women in particular. They say:
In older women gamma-tocopherol and gamma-tocopherol:alpha-tocopherol ratios were directly related to indices of obesity. In young men alpha- and gamma-tocopherols were directly correlated with obesity, but gamma-tocopherol:alpha-tocopherol ratio was not.
We mentioned this finding to a physician we know. He didn’t have an immediate explanation for why gamma-tocopherol might be related to BMI, but he pointed out that vitamin E is fat-soluble, so it could just be that if you’re overweight, you have more body fat, which can store more of these fat-soluble vitamins. More fat, more gamma-tocopherol stored in your body, and more in your serum.
This is plausible — anything that gets stored in fat will probably show up in higher quantities in the bodies of people with more body fat. And this is a major possible confounder in general for this kind of big correlational analysis, keep it in mind for future posts (in case you’re wondering, copper and other metals in general are not fat-soluble).
But one strike against the idea that fat accumulation is driving the relationship with BMI is that the correlation between BMI and other tocopherols is weaker.
The relationship between BMI and serum alpha-tocopherol is also significant, but the magnitude of the relationship is much smaller.
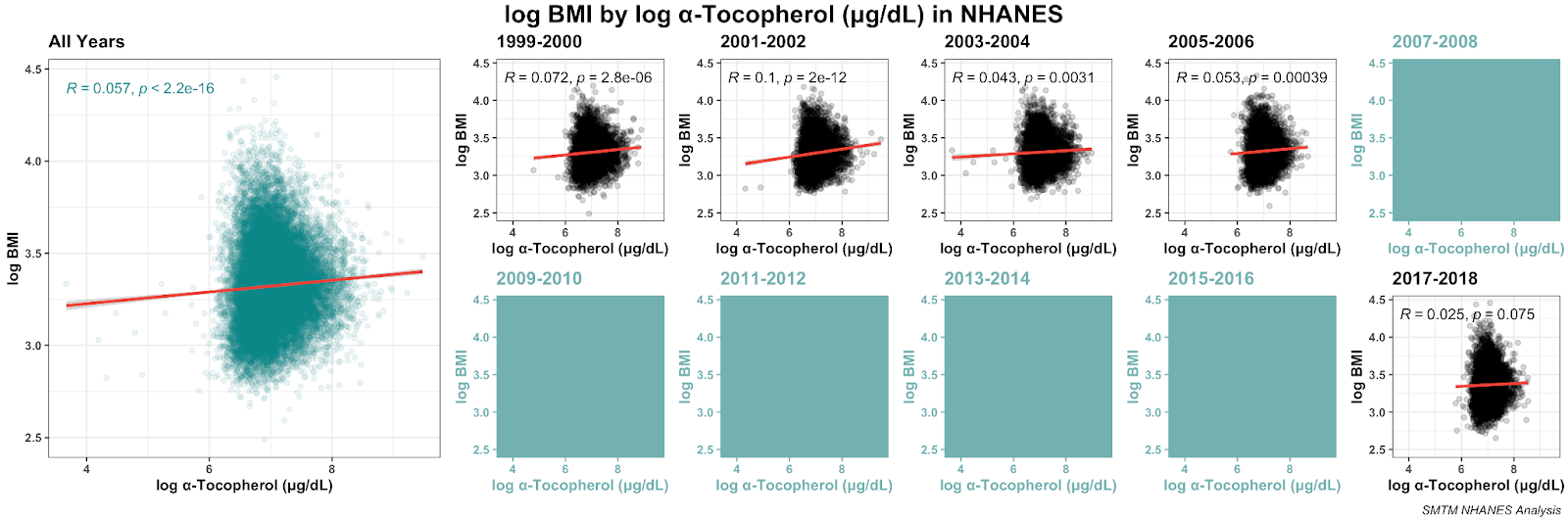
Here’s the distribution of serum alpha-tocopherol in those years:
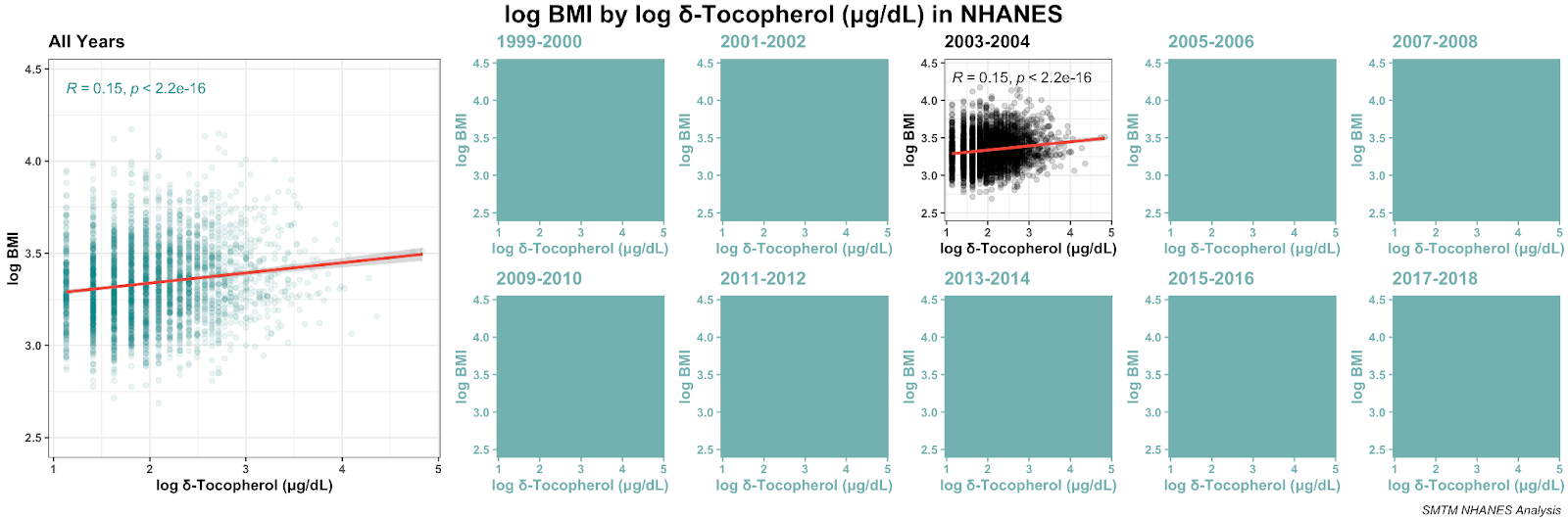
We also happen to have one year of data for delta-tocopherol, so why not. It also has a bit of a relationship with BMI, stronger than alpha but weaker than gamma.
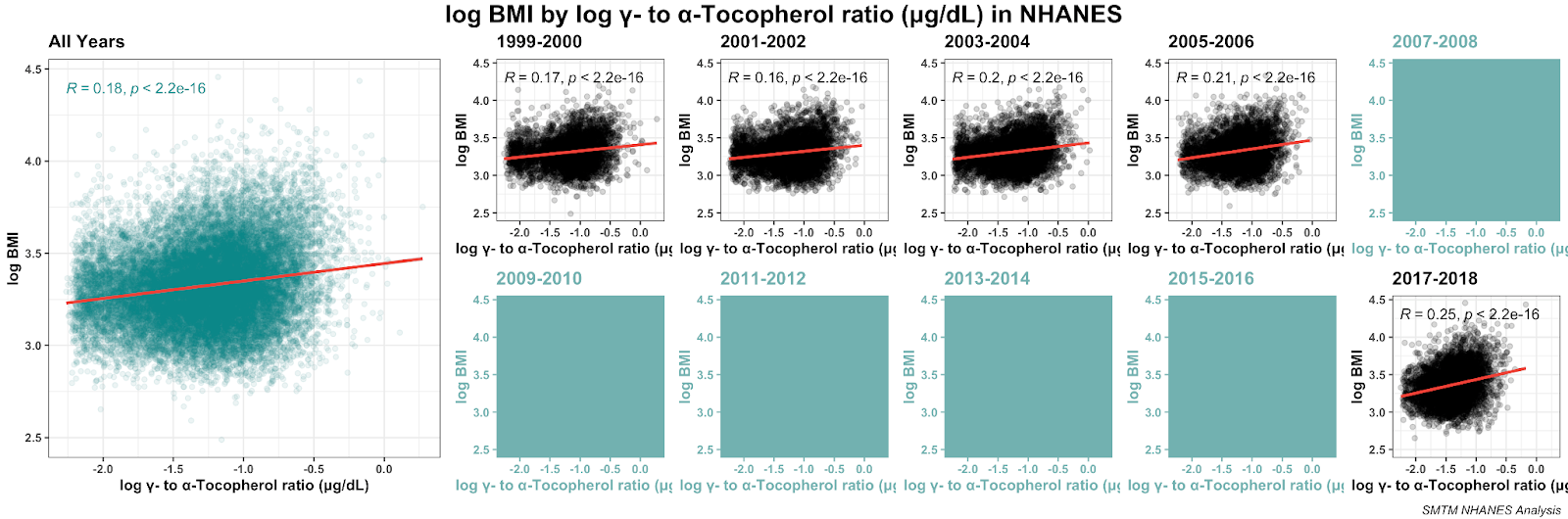
Some of the literature suggests that the ratio of gamma- to alpha-tocopherol is important, and yeah, the ratio is indeed correlated with BMI. Again we can plot it. We removed two weird outliers (their ratios are about 5x higher than everyone else’s) to make the plot a little cleaner, but removing them doesn’t make a difference to the overall correlation:
Here’s the distribution for those ratios in these years:
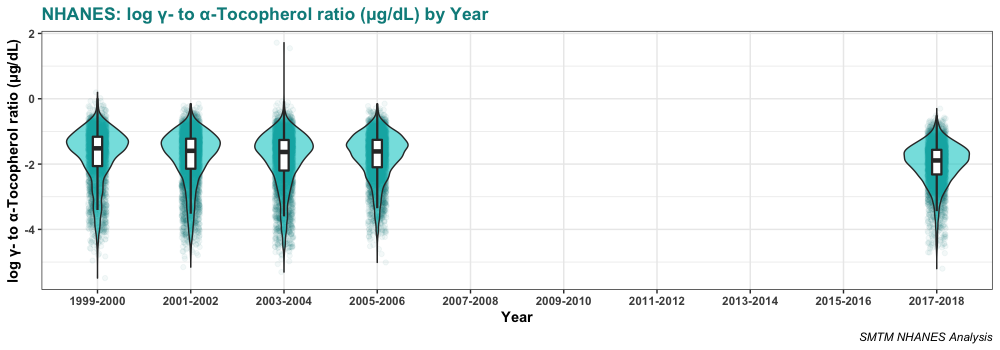
Again we want to note that the average ratio is going down over time. If the gamma-tocopherol:alpha-tocopherol ratio were causing obesity, you’d expect it to go up over time.
While the gamma-tocopherol:alpha-tocopherol ratio is significantly correlated with BMI, this correlation is smaller than the correlation between BMI and gamma-tocopherol by itself. Does the ratio actually add anything to the picture?
Looks like yes. In a multiple regression predicting log BMI, both alpha- and gamma-tocopherol are significant predictors, and so is their interaction, all p-values extremely small. A model consisting of just alpha-tocopherol, gamma-tocopherol, and their interaction predicts about 7% of the variance in log BMI.
However, in this model both alpha- AND gamma-tocopherol have positive coefficients, meaning a higher serum level of either tocopherol predicts higher BMI. And their interaction has a negative coefficient, meaning that they are less than the sum of their parts.
That’s kind of weird. Here’s our best interpretation. Gamma-tocopherol is clearly associated with BMI. But alpha-tocopherol is barely associated with BMI at all, and while the correlation is sometimes significant, it’s much more tenuous. In a multiple regression, the interaction is negative. This means that the association between gamma-tocopherol and BMI is weaker as people have more alpha-tocopherol in their serum, so this is at least consistent with the idea that alpha-tocopherol has a protective effect against bad effects of gamma-tocopherol (more on causal inference in a moment).
If we include the gender effect we mentioned above, all these variables are still significant, all the interactions that were previously significant are still significant, and the alpha-tocopherol x gamma-tocopherol x gender three-way interaction is also significant. This model explains about 8% of the variance in BMI.

Interpretation
Ok, what are we to make of this?
This review from 2001 is a good starting place for anyone interested in reading more about “the bioavailability, metabolism, chemistry, and nonantioxidant activities of γ-tocopherol and epidemiologic data concerning the relation between γ-tocopherol and cardiovascular disease and cancer.” Here are some quotes that we found especially relevant:
γ-Tocopherol is the major form of vitamin E in many plant seeds and in the US diet, but has drawn little attention compared with α-tocopherol, the predominant form of vitamin E in tissues and the primary form in supplements. However, recent studies indicate that γ-tocopherol may be important to human health and that it possesses unique features that distinguish it from α-tocopherol.
γ-Tocopherol is often the most prevalent form of vitamin E in plant seeds and in products derived from them (10). Vegetable oils such as corn, soybean, and sesame, and nuts such as walnuts, pecans, and peanuts are rich sources of γ-tocopherol (10). Because of the widespread use of these plant products, γ-tocopherol represents ≈70% of the vitamin E consumed in the typical US diet (10).
In humans, plasma α-tocopherol concentrations are generally 4–10 times higher than those of γ-tocopherol (13).
We can compare this to some other sources as well. Here’s an old USDA poster on nuts and seeds as sources of both alpha- and gamma-tocopherol. According to this source, “the highest sources of alpha-tocopherol in nuts and seeds are sunflower seeds, almonds/almond butter, hazelnuts, and pine nuts. The highest sources of gamma-tocopherol are black walnuts, sesame seeds, pecans, pistachios, English walnuts, flaxseed, and pumpkin seeds.”
This page from the NIH says, “most vitamin E in American diets is in the form of gamma-tocopherol from soybean, canola, corn, and other vegetable oils and food products.”
The Wikipedia page for “Tocopherol” says,
α-tocopherol is the main source found in supplements and in the European diet, where the main dietary sources are olive and sunflower oils, while γ-tocopherol is the most common form in the American diet due to a higher intake of soybean and corn oil.
As far as we can tell, animal fats seem to be high in alpha-tocopherol, but we can’t find great sources on this. If you find one that looks reliable, please let us know.
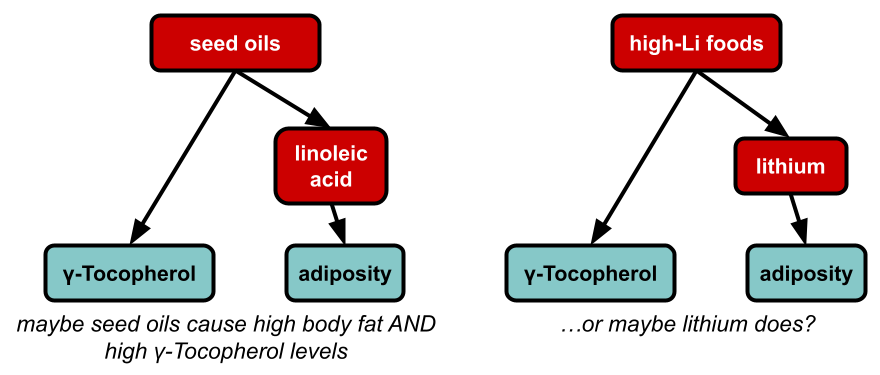
This seems like remarkably good news for the seed oil theorists, who think that the obesity epidemic comes from the fact that we started using lots of new food oils derived from plant seeds, like soybean, corn, and canola oils. We’ve previously looked into seed oils as an explanation and didn’t find much evidence in favor of the idea, but we don’t think it’s totally implausible.
Seed oil theorists usually pin the blame on linoleic acid as the reason seed oils might be making people obese, but maybe they’re wrong. Maybe it’s actually tocopherol ratios. Maybe they picked the right theory for the wrong reasons — it happens. Or maybe gamma-tocopherol is only correlated with BMI because it’s a proxy measure for how much seed oil you’re eating. Causal inference is hard y’all.
We don’t think that gamma-tocopherol causes obesity. If it did, then you would expect gamma-tocopherol levels (or the ratio with alpha-tocopherol) to increase over time. Instead, they have slightly decreased. And just in general, we don’t think it fits well with the shape of the obesity epidemic. There are big differences in obesity rates between different professions, and we don’t think auto mechanics are somehow getting hugely more gamma-tocopherol in their diet than teachers.
More likely, having more body fat makes you retain more gamma-tocopherol for an unrelated reason, possibly because vitamin E is a fat-soluble vitamin.

You could maybe test this by looking at the serum tocopherol levels of a group that was gaining weight for a known reason, for example people who are starting olanzapine, an antipsychotic that often causes weight gain. If they gained weight but their tocopherol levels didn’t change, that would suggest that adiposity doesn’t have a direct effect on tocopherol levels.
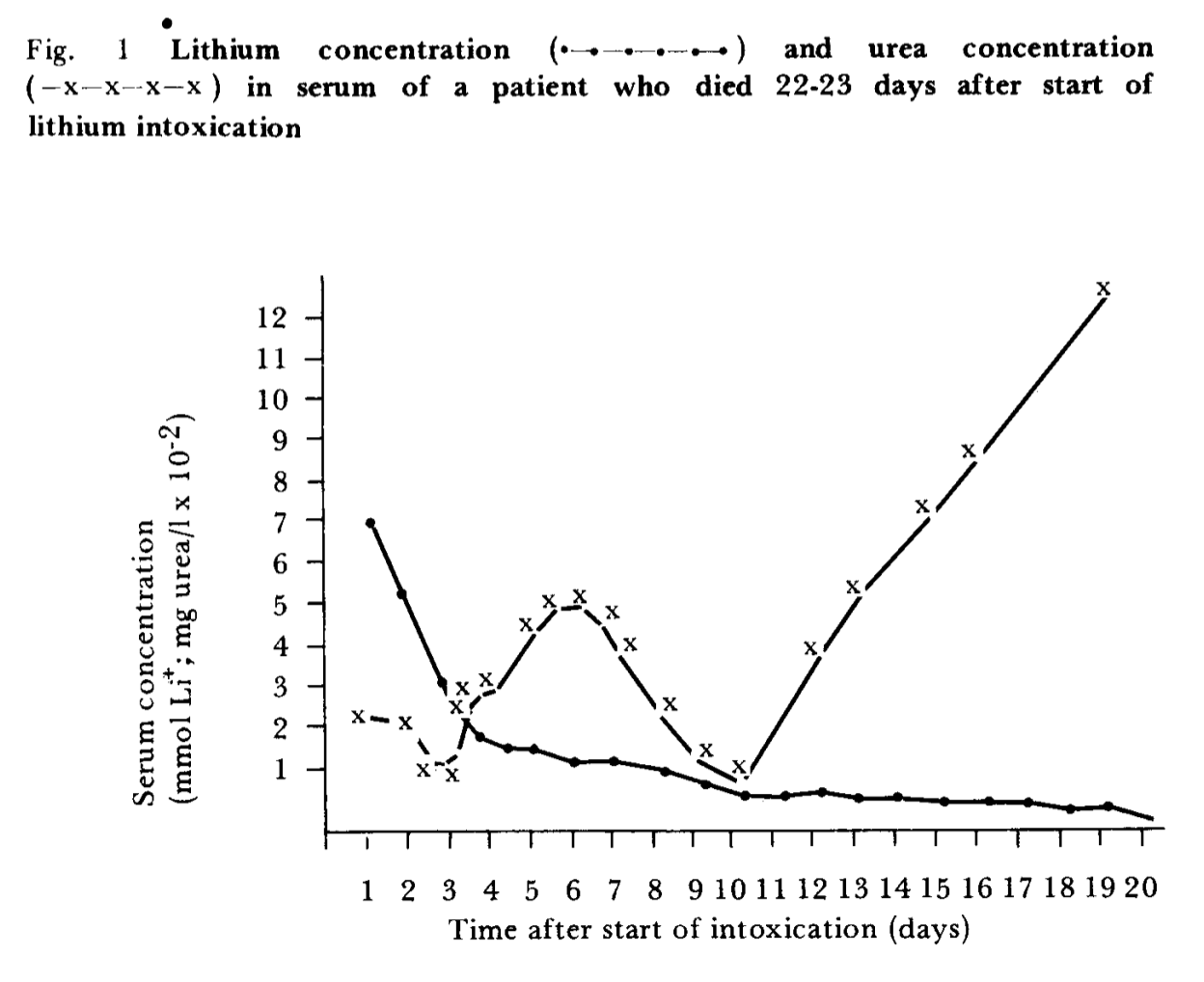
Or, the thing that is really causing the obesity epidemic also happens to increase gamma-tocopherol for unrelated reasons. For example, if linoleic acid is the cause of the obesity epidemic, then you would expect people who are obese to have high levels of gamma-tocopherol, because foods that are high in linoleic acid (like soybean oil) also tend to be high in gamma-tocopherol. Or if lithium is the cause of the obesity epidemic, then maybe people who are obese would have high levels of gamma-tocopherol, if the kinds of foods that accumulate lithium (perhaps pumpkin seeds?) also tend to be high in gamma-tocopherol.
But that said, it would be pretty straightforward to try to eat a diet that’s high in alpha-tocopherol and/or low in gamma-tocopherol, and see what happened. If you started losing weight immediately, that would be quite striking, and you could maybe back it up with blood tests to measure your serum tocopherol levels.
So it’s worth considering if anyone has tried a high-alpha / low-gamma tocopherol (HALGT?) diet already.
The ketogenic diet seems like it might sometimes be a HALGT diet, depending on the fats you use. If you focused on olive oil, almonds, and animal fats (for example), you would be getting a lot of alpha-tocopherol and not much gamma-tocopherol. But if you focused on fat from walnuts, peanut butter, and corn oil, you would be getting the opposite. This would kind of fit with the picture of how keto seems to have amazing effects for some people and basically no effect for others.

It would certainly fit with the Shangri-La diet. This is a “diet” that basically just involves taking two tablespoons of olive oil every morning. Seth Roberts, who developed this approach, claimed that this was enough to totally kill his appetite and make him lose weight, and this is backed up by some other anecdotes (here’s one example).
Well, olive oil contains mostly alpha-tocopherol. Taking olive oil every morning would definitely increase your alpha-tocopherol intake, and depending on how much alpha-tocopherol and gamma-tocopherol you’re getting from the rest of your diet, this might be enough to change your ratio or whatever. But someone who was getting a lot of gamma-tocopherol from other sources might just have their gamma-tocopherol wipe out the alpha-tocopherol from the olive oil, and this might explain why the Shangri-La diet works for some people but not for other people.
What about the croissant diet? We’re not sure. This diet involves getting a lot of fat from butter and other animal sources, so if it’s true that these are high in alpha-tocopherol, then the croissant diet might be a HALGT diet. It would also depend on how much gamma-tocopherol is in the other foods from this diet. The same logic would probably apply for many carnivore-style diets.
Ok, how about the potato diet? Potatoes contain a little vitamin E, and most sources strongly hint that this is mostly or entirely alpha-tocopherol. For example, the USDA says that 100g of potato contains 0.01 mg Vitamin E as alpha-tocopherol, and 0 mg of beta-, gamma-, and delta-tocopherol. Similarly, this U Rochester page says that potatoes contain 0.04 mg “Vitamin E (alpha-tocopherol)” per “1 Potato large”.
This is not much vitamin E — the recommended dose is 15 mg a day. So if anything, the potato diet looks more like a vitamin E near-elimination diet. Maybe the elimination is the real factor, and it lets your body re-balance your tocopherol levels? But aside from that elimination, you are getting just a little vitamin E as alpha-tocopherol. And depending on the cooking oil you use, you might be getting more or less. This perspective suggests that the potato diet might work better with high-alpha-tocopherol fats like olive or sunflower oil and would work worse, or possibly not at all, with high-gamma-tocopherol fats like corn or soybean oil. That at least is an empirical prediction (assuming all these measurements of the tocopherol levels in foods are at all accurate).
Even if the HALGT hypothesis is entirely accurate, though, there will always be lots of complications. Most of the variation in BMI between different people remains genetic. Tocopherols are destroyed by exposure to high temperatures, so the way you cook your foods and the methods used to process your cooking oils might make a huge difference. So maybe cold olive oil has 10x the effect of olive oil that was heated and used to cook food. If it isn’t this difference, it’s inevitably something else. So even if the real answer to obesity is as simple as the ratio of your alpha- to gamma-tocopherol, expect it to also be at least this complicated.
So color us still skeptical but, there’s some evidence pointing in this direction.