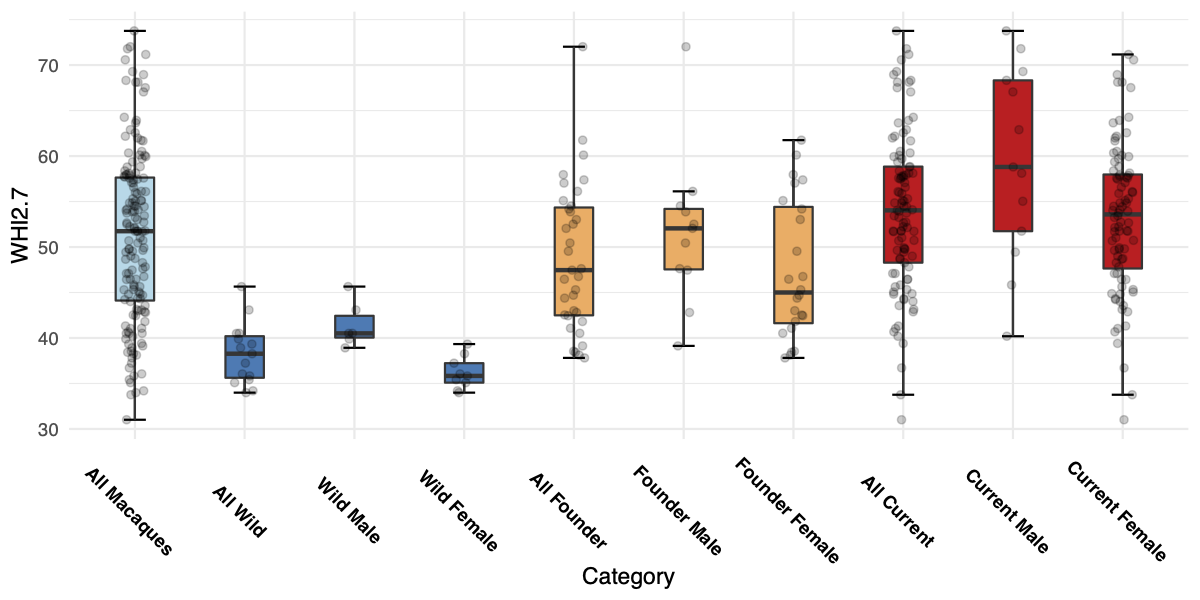
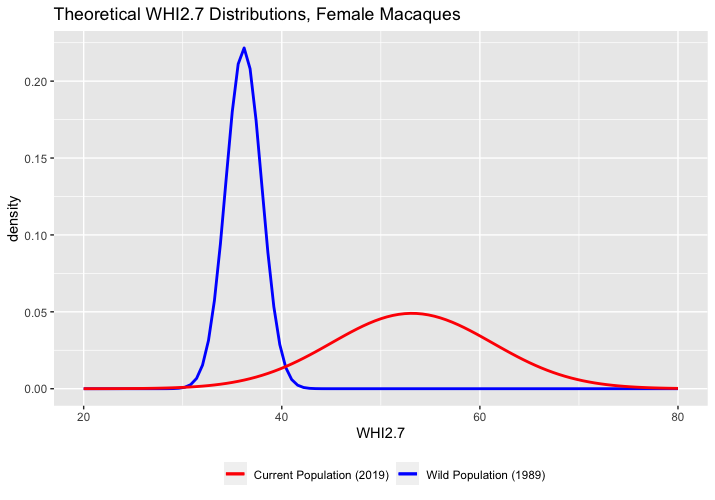
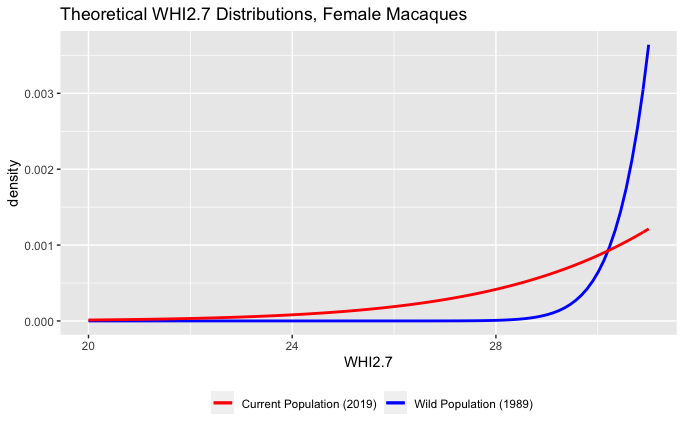
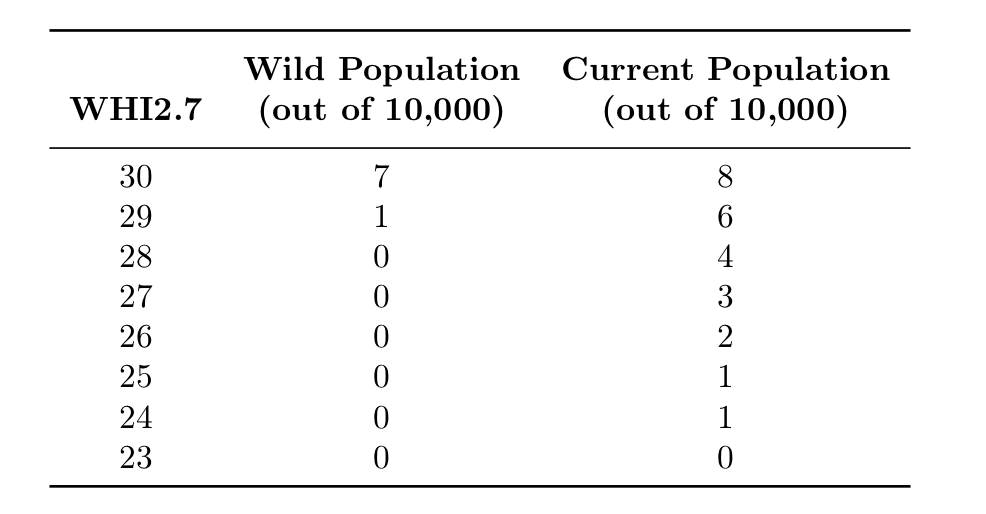
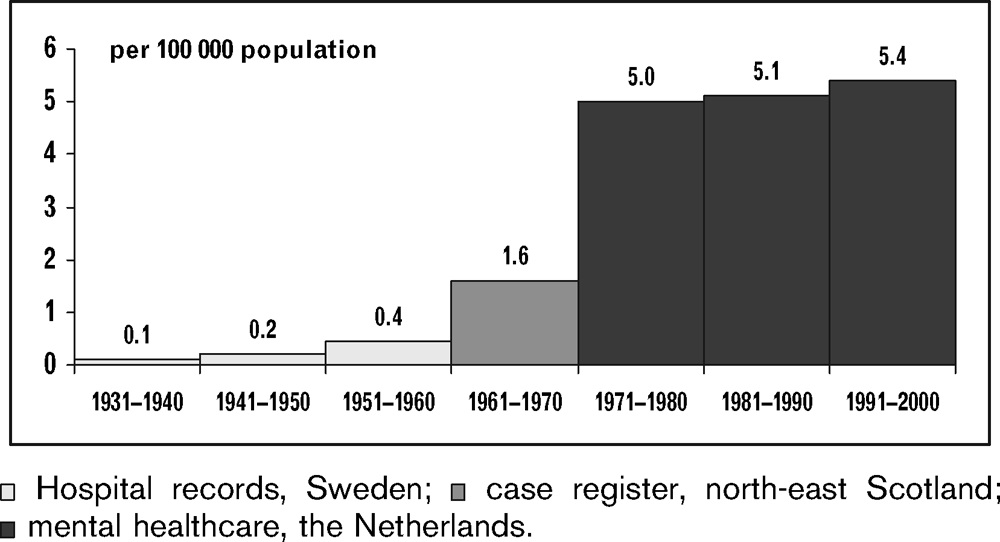
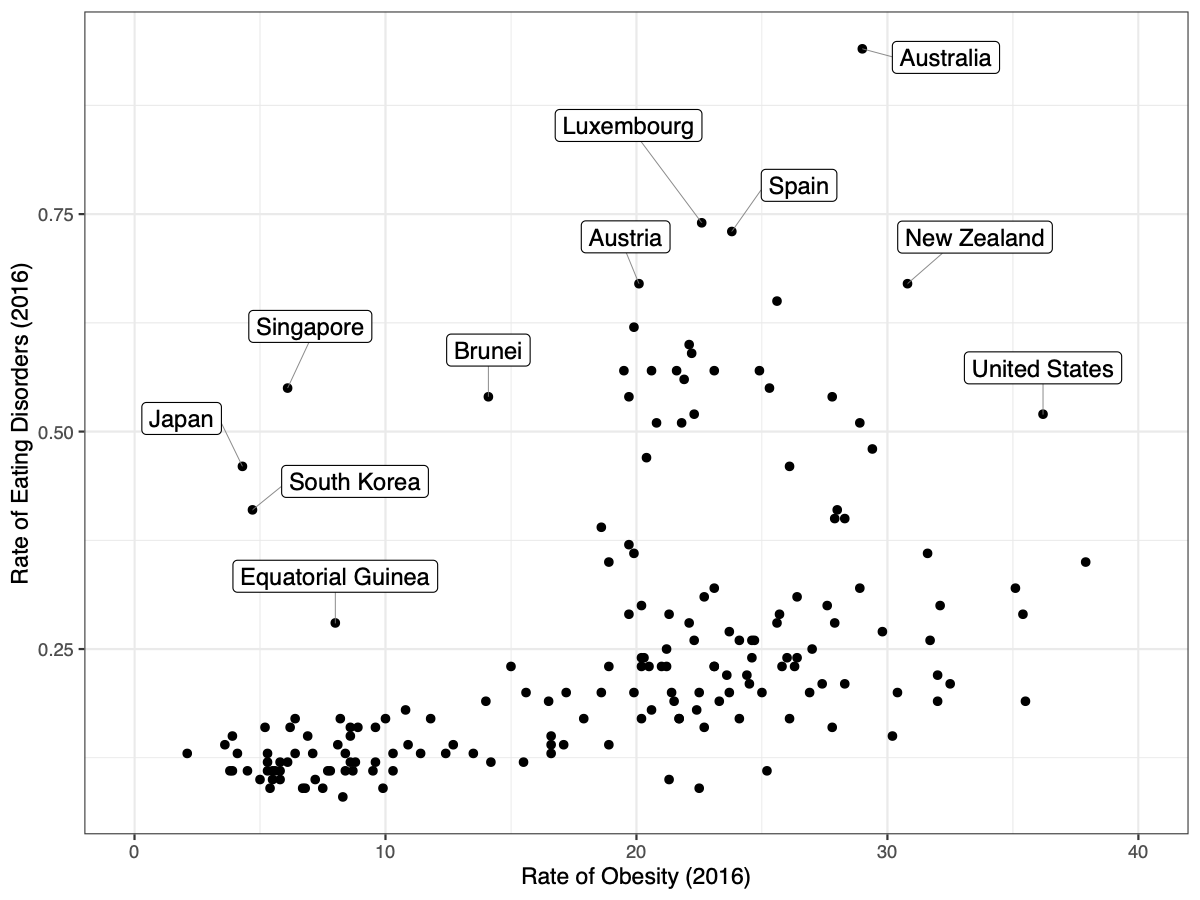
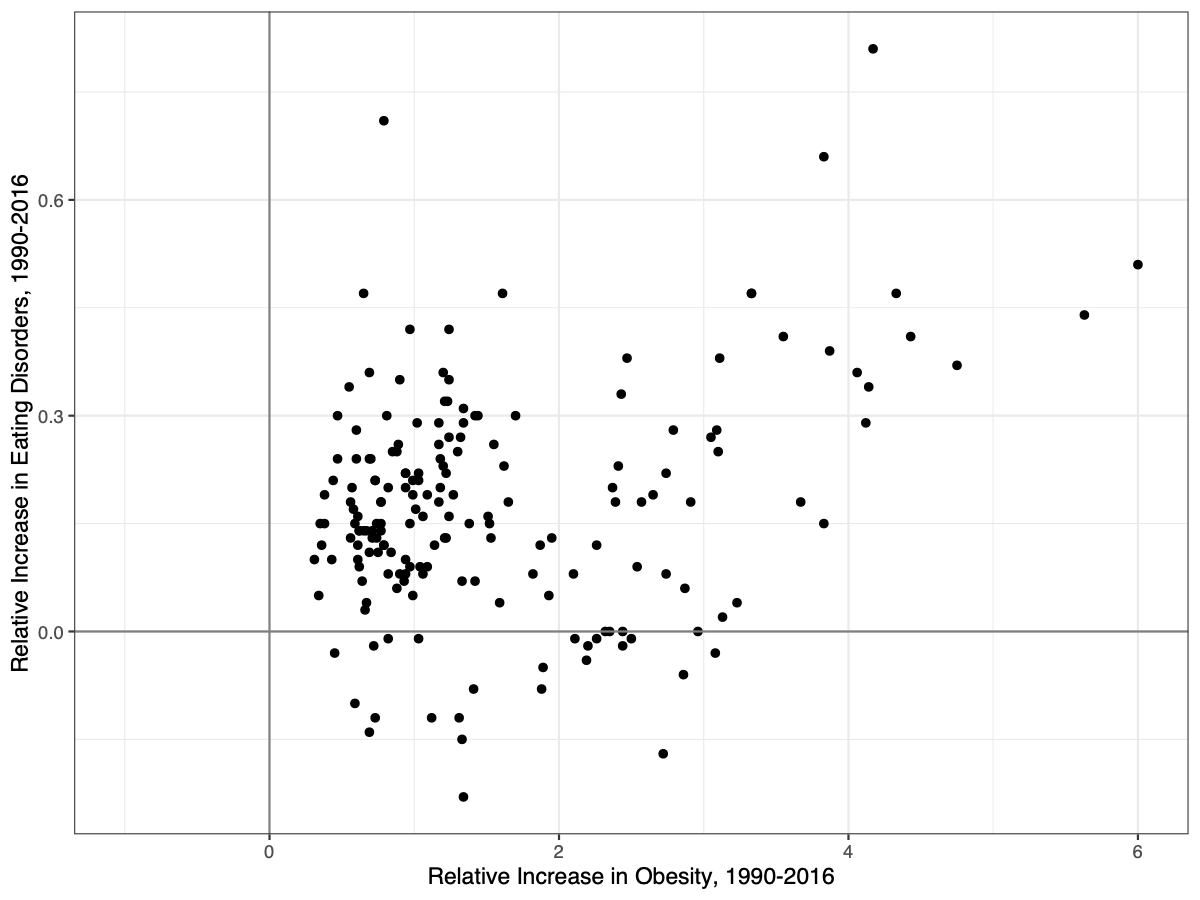
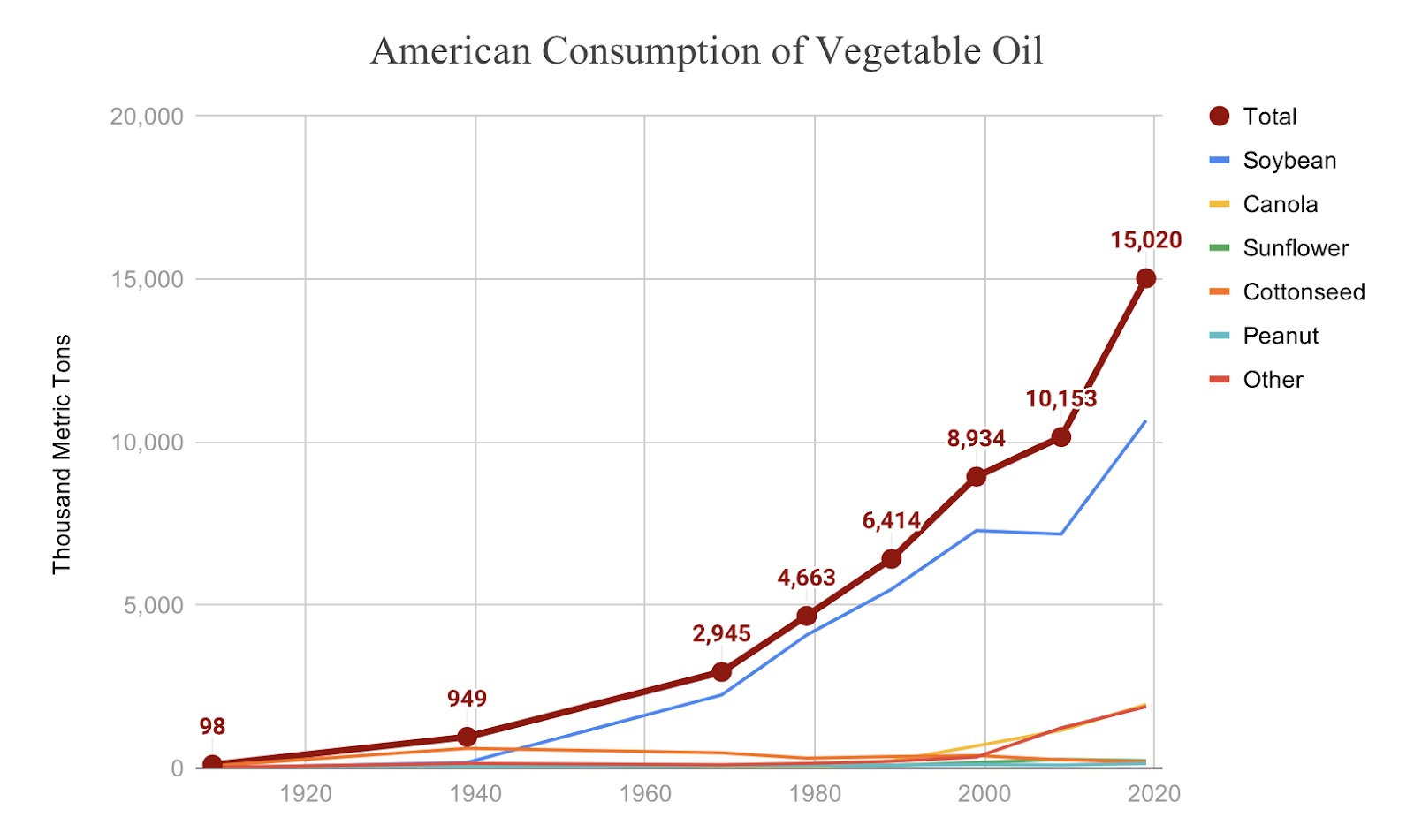
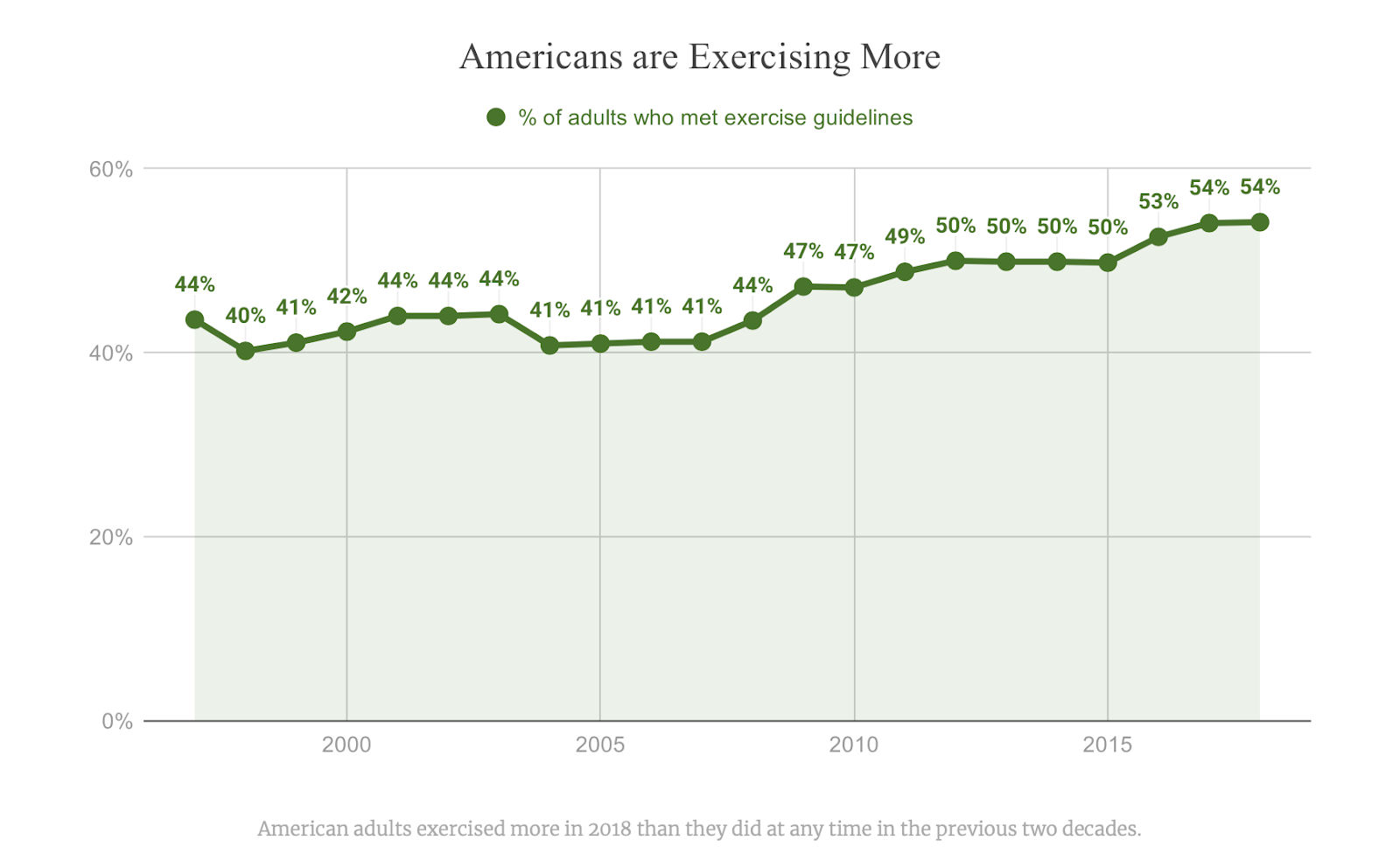
[PART I – MYSTERIES]
[PART II – CURRENT THEORIES OF OBESITY ARE INADEQUATE]
[PART III – ENVIRONMENTAL CONTAMINANTS]
[INTERLUDE A – CICO KILLER, QU’EST-CE QUE C’EST?]
[PART IV – CRITERIA]
[PART V – LIVESTOCK ANTIBIOTICS]
[INTERLUDE B – THE NUTRIENT SLUDGE DIET]
[PART VI – PFAS]
[PART VII – LITHIUM]
[INTERLUDE C – HIGHLIGHTS FROM THE REDDIT COMMENTS]
[INTERLUDE D – GLYPHOSATE (AKA THE ACTIVE INGREDIENT IN ROUNDUP)]
[INTERLUDE E – BAD SEEDS]
[PART VIII – PARADOXICAL REACTIONS]
[PART IX – ANOREXIA IN ANIMALS]
[INTERLUDE F – DEMOGRAPHICS]
[INTERLUDE G – Li+]
[INTERLUDE H – WELL WELL WELL]
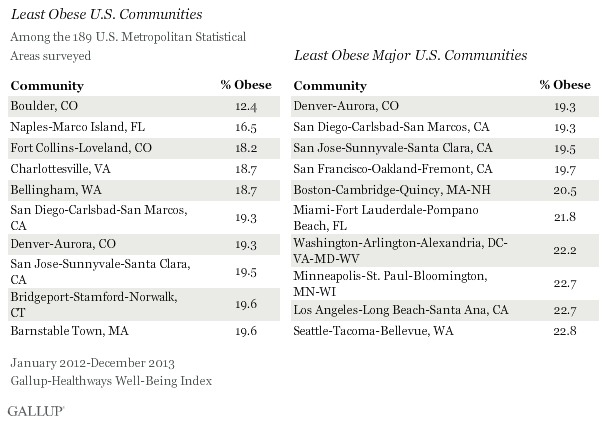
[INTERLUDE I – THE FATTEST CITIES IN THE LAND]
Assuming you take our main thesis seriously — that obesity is the result of environmental contaminants — what should you do about it?
Our suggestions are very prosaic: Be nice to yourself. Eat mostly what you want. Trust your instincts.
Diet and exercise won’t cure obesity, but this is actually good news for diet and exercise. You don’t need to put the dream of losing weight on their shoulders, and you can focus on their actual benefits instead. You should focus on your diet — not to get thin, but to make sure that you have enough energy to do everything you want to do in life. This means eating enough and making sure you get what you need. You should exercise — not to slim down, but to gain strength and energy, and you shouldn’t get discouraged when you don’t drop 50 lbs fast.
Don’t be mean to fat people. If you’re fat, don’t be mean to yourself about it. Don’t be a dick.

And this doesn’t apply to most of our readers, of course, but just in general — we gotta stop spending money on circular nutrition research. It’s clearly not going anywhere. Other theories of obesity don’t engage with the observations that are out there about the obesity epidemic, and try to explain the wrong thing.
Most theories focus on the dynamics of individual weight loss, under the assumption that obesity is the result of the normal mechanics of eating, exercise, weight loss, and weight gain. But we think that the dynamics of individual weight loss have almost nothing to do with the real question, which is why obesity rates are so much higher now than they were in the 1970s, and the rest of human history. Individuals can gain or lose 15-20 lbs from their set point, but this is messing around within the range of control — we only care about the set point.
Let’s say it’s 50 °F outside. If your thermostat is set to 72 °F and you open the door, your house’s temperature will drop at first and then will go back up to the set point of 72 °F. If your thermostat is set to 110 °F and you open the door, your house’s temperature will drop at first and then will go back up to the set point of 110 °F (assuming your furnace is strong enough).
This is a standard feature of how homeostatic systems respond to major disturbances — the controlled value swings around for a bit until the system can get it back under control, and send it back to the set point. So all the diet and exercise studies we’ve done over the last 50 years have just been an exercise in who can create the biggest, most jarring disturbance — but the lipostat always finds a way to bring your weight back where it wants it.
So all these “punch the control system as hard as we can” studies don’t tell us anything about why the thermostat is set to 110 °F in the first place, which is what we’re really interested in.
Get It Outta Me
Bestselling nutrition books usually have this part where they tell you what you should do differently to lose weight and stay lean. Many of you are probably looking forward to us making a recommendation like this. We hate to buck the trend, but we don’t think there’s much you can do to keep from becoming obese, and not much you can do to drop pounds if you’re already overweight.
We gotta emphasize just how pervasive the obesity epidemic really is. Some people do lose lots of weight on occasion, it’s true, but in pretty much every group of people everywhere in the world, obesity rates just go up, up, up. We’ll return to our favorite quote from The Lancet:
“Unlike other major causes of preventable death and disability, such as tobacco use, injuries, and infectious diseases, there are no exemplar populations in which the obesity epidemic has been reversed by public health measures.”
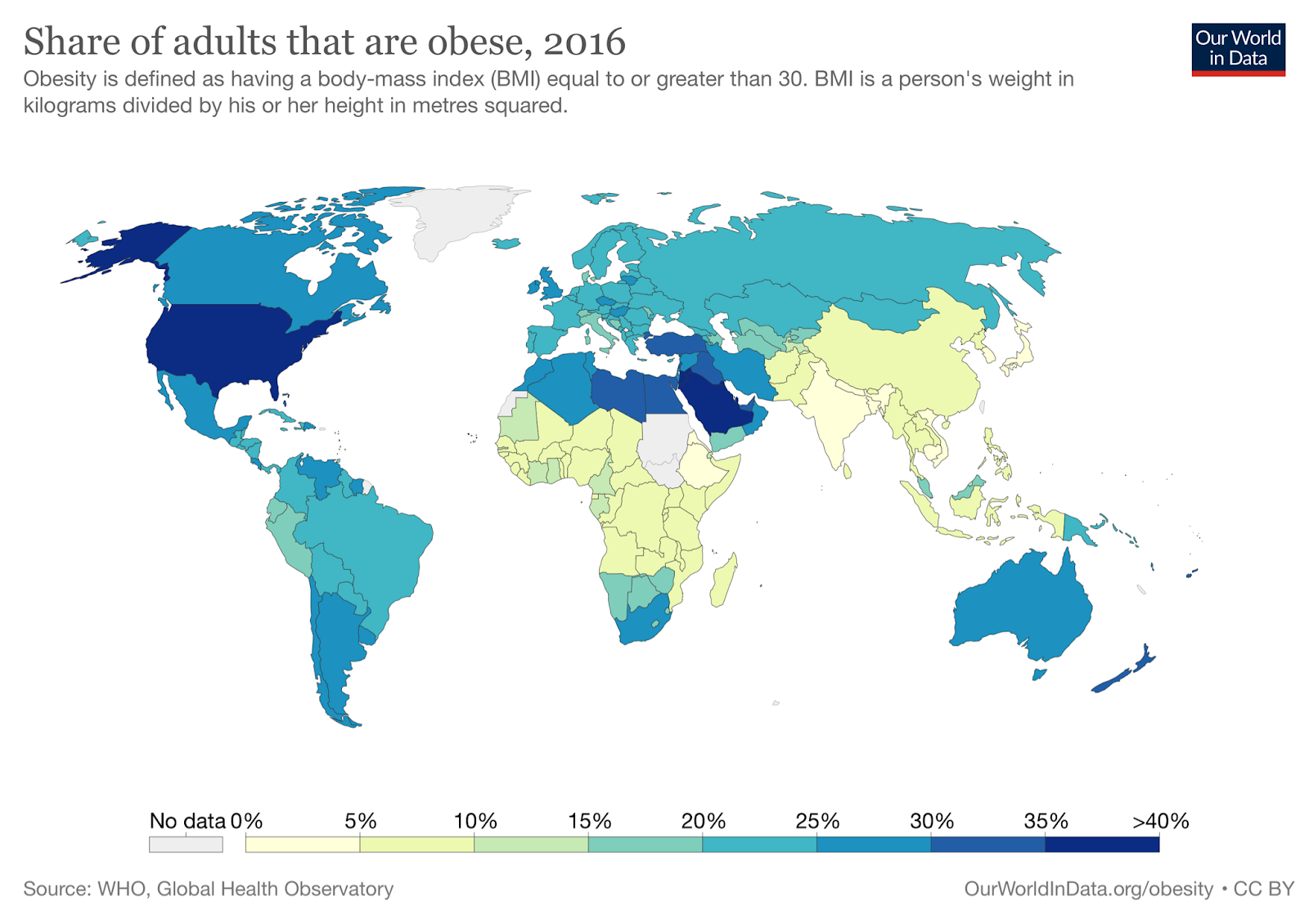
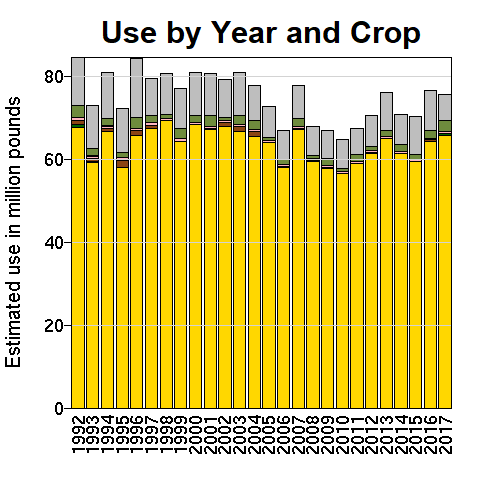
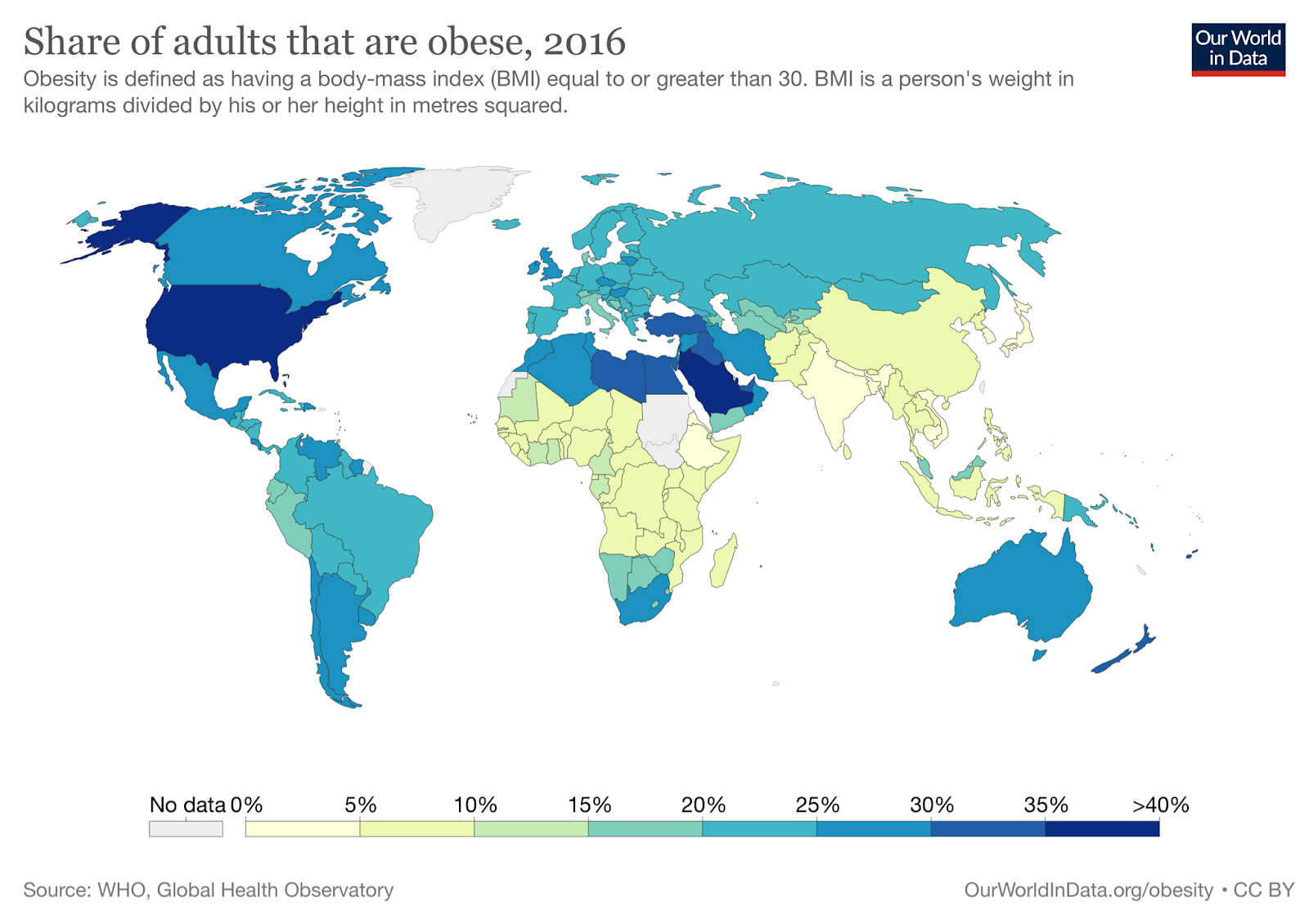
The nonprofit ourworldindata.org has data from the WHO covering obesity rates in almost every country in the world from 1975 to 2016. In every country in this dataset, the obesity rate either stayed the same or increased every single year from 1975 to 2016. There is not one example of obesity rates declining for even a single country in a single year. Countries like Japan and Vietnam are some of the leanest countries in the world (about 4% and 2% obese, respectively), but in this dataset at least, even these super-lean countries don’t see even a single year where their obesity rates decline.
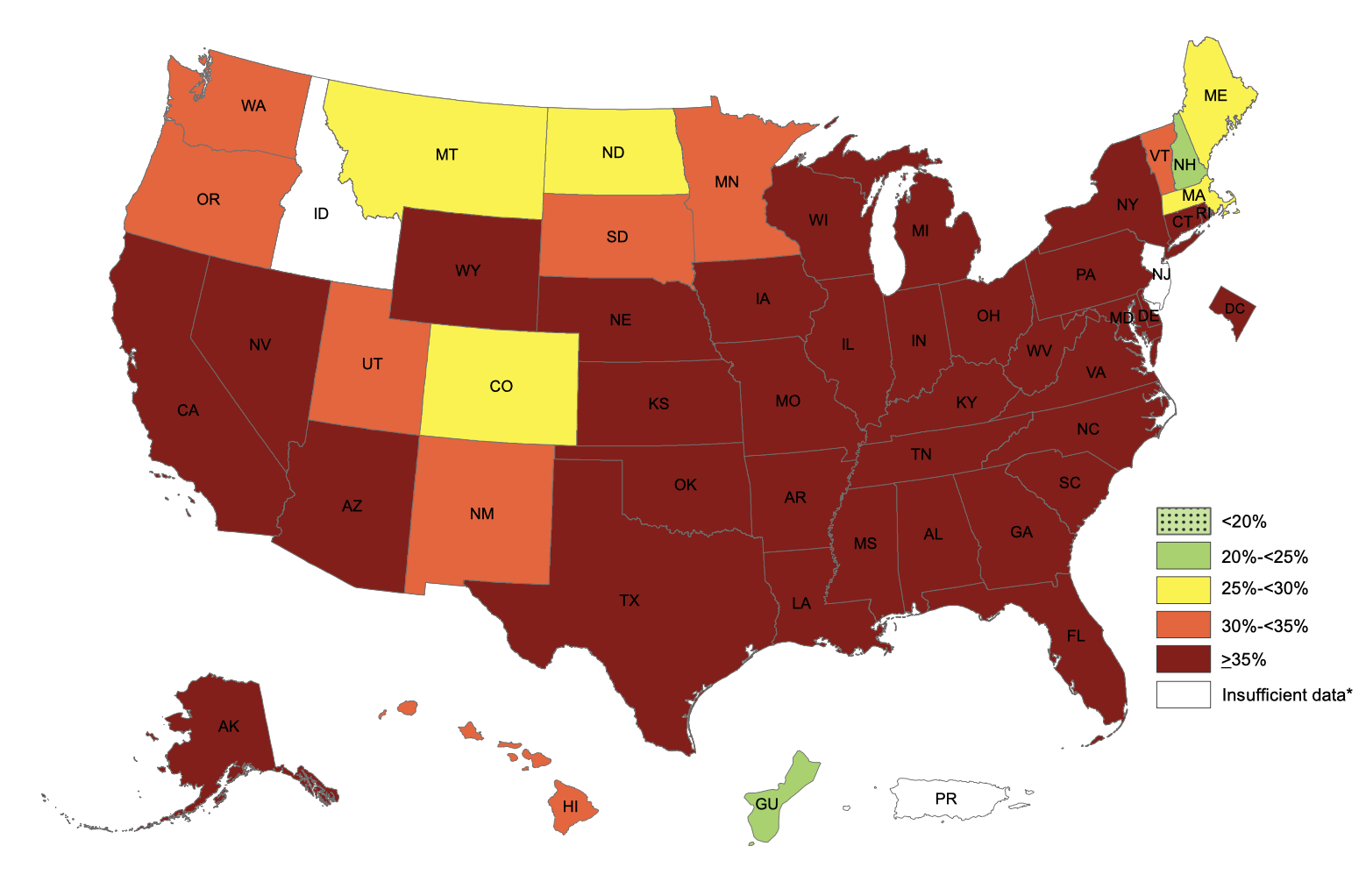
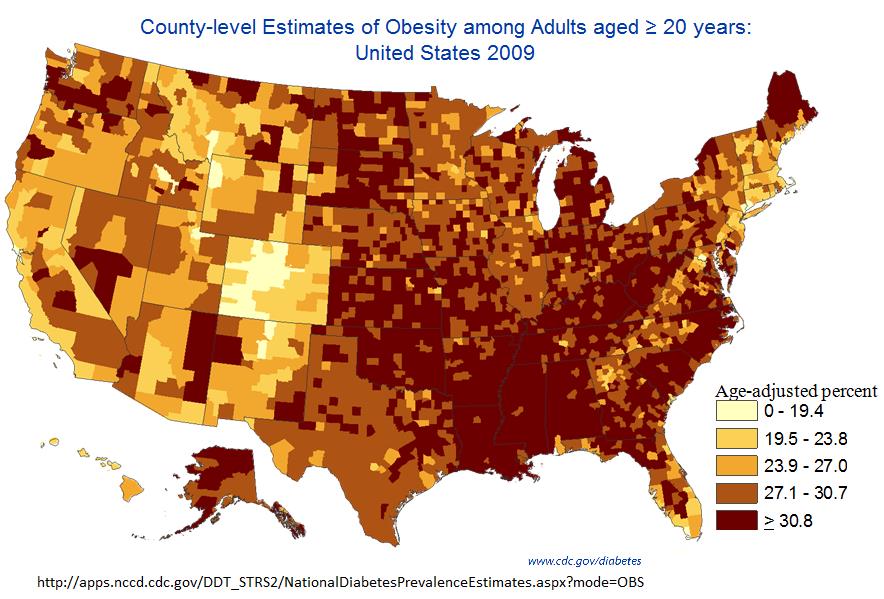
We see the same trend even for smaller-scale data. The Institute for Health Metrics and Evaluation (IHME) has a dataset of county-level obesity data from 2001 to 2011, which is publicly available on their website. Using this we can look at obesity rates across the United States, and we can see how much obesity rates have changed in each county between 2001 and 2011. We see that between 2001 and 2011, obesity rates decreased in zero counties, stayed the same in zero counties, and increased in 3,143 out of 3,143 counties and county equivalents in the United States.

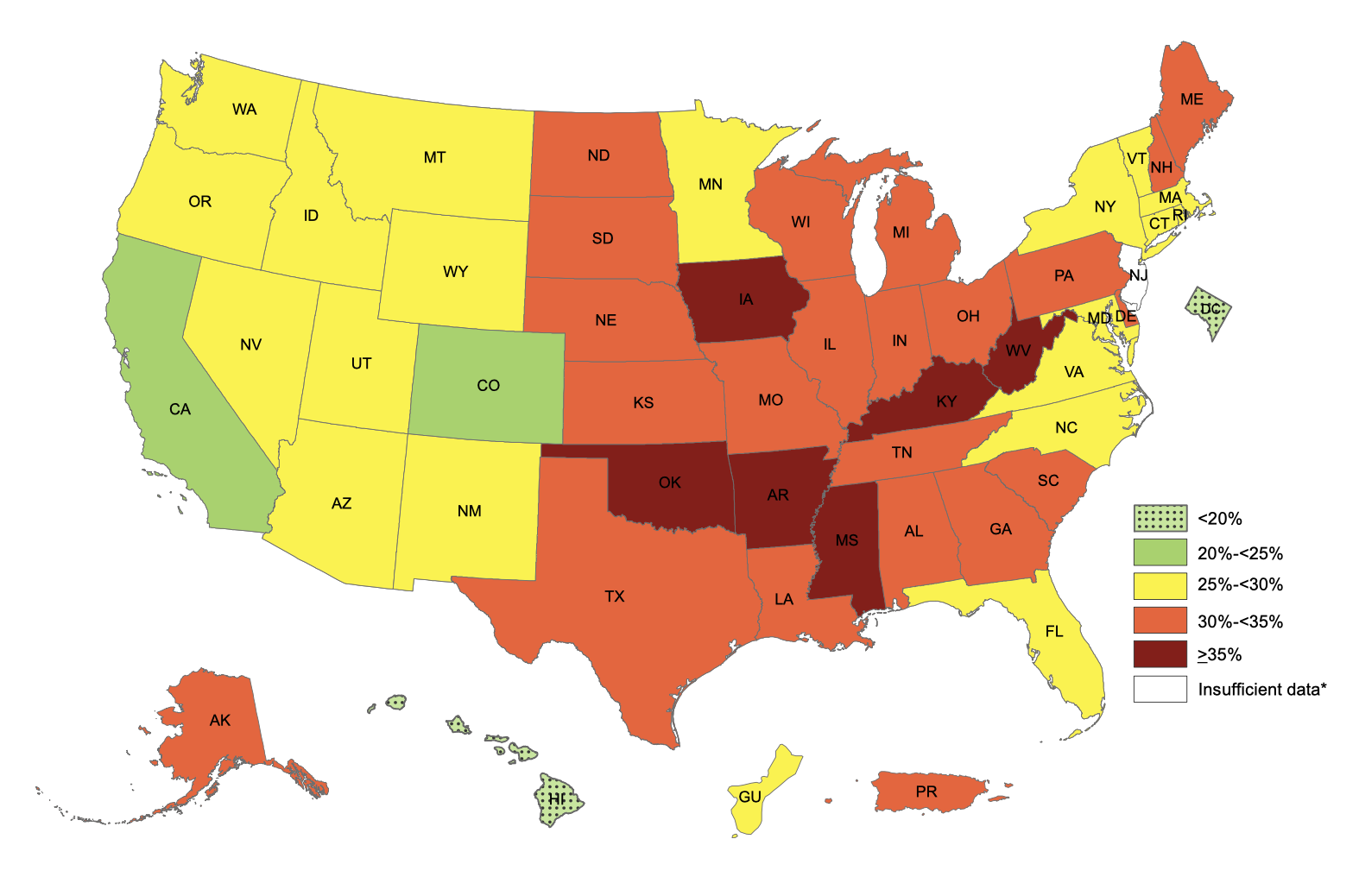
The smallest increase between 2001 and 2011 was in Eagle County, Colorado, where obesity rates went from 20.0% in 2001 to 21.5% in 2011, an increase of 1.5%. You’ll notice that this is Colorado once again, and it turns out that the five counties with the smallest increase from 2001 to 2011 are all in Colorado. Of the 25 counties with the smallest increase, 13 are in Colorado. The take-home here is that Colorado really is special.
If we zoom in a little further on these data, we can find ONE case of obesity rates declining — they went from 22.7% in 2009 to 22.4% in 2011 in Fairfax City, Virginia, a drop of 0.3%. There were also two counties where rates stayed the same 2009-2011. But this is one county with rates going down, two staying the same, and 3,140 going up. If population-level reversals are this tiny and this rare, it’s hard to imagine that there is much an individual can do to change their own weight.
But that said, here are a few ideas, approximately in order from least extreme to most extreme.
First off, there are a few things that won’t change how many contaminants you’re exposed to, but that may have an impact on your weight anyways.
1. — The first is that you can put on more muscle mass. This won’t affect your weight as it appears on the scale, but it does often seem to affect people’s body composition. The lipostat pays attention to how much fat you have, but it also seems to pay some attention to how much you literally weigh (see these studies in mice, and this recent extension in humans). So if you gain muscle mass, you may lose fat mass. For advice on how to gain muscle mass, please see the internet.
2. — The second is that you could consider getting gastric bypass or a similar, related surgery. Our understanding is that these procedures are very effective at causing weight loss in many cases. However, they are pretty dangerous — this is still a surgical procedure, and so inherently comes with a risk of death and other serious complications. If you consider this option please take it very seriously, consult with your doctor, etc.
Many of you, however, are not just interested in weight loss, or are interested in weight loss along with reducing how many mystery chemicals you’re exposed to — “You stupid kids I don’t want to lose weight I want to get these contaminants out of my body!!!” So here’s a list of steps you could take to reduce your exposure and possibly lose weight, again approximately in order from least extreme to most extreme.
1. — The first thing you should consider is eating more whole foods and/or avoiding highly processed foods. This is pretty standard health advice — we think it’s relevant because it seems pretty clear that food products tend to pick up more contaminants with every step of transportation, packaging, and processing, so eating local, unpackaged, and unprocessed foods should reduce your exposure to most contaminants.
2. — The second thing you can do is try to eat fewer animal products. Vegetarians and vegans do seem to be slightly leaner than average, but the real reason we recommend this is that we expect many contaminants will bioaccumulate, and so it’s likely that whatever the contaminant, animal products will generally contain more than plants will. So this may not help, but it’s a good bet.
3. — The third thing is you can think about changing careers and switching to a leaner job. Career is a big source of variance in obesity rates, so if you have a job in a high-obesity profession like truck driver or mechanic, consider switching to a job in a low-obesity profession like teacher or surveyor. For a sense of what careers are high- and low-obesity, check out this paper about obesity by occupation in Washington State and this paper about obesity by occupation in US workers. If you are already in a pretty lean career, then ignore this one.
We think this goes double if you’re in a profession where you’re working with lithium grease directly, or even around lithium grease. Do what you can to stay away from the stuff.
4. — The fourth thing you can consider is changing where you live. The simplest is to change where you live locally — stay in the same area, but move to a different house or apartment. This one is tricky, and sort of a shot in the dark. How will you know if you are moving to a more or less-contaminated house? But if you suspect your house is high in contaminants, it might be worth moving. If you find specific contaminants especially concerning, you can try having your local water tested for them.
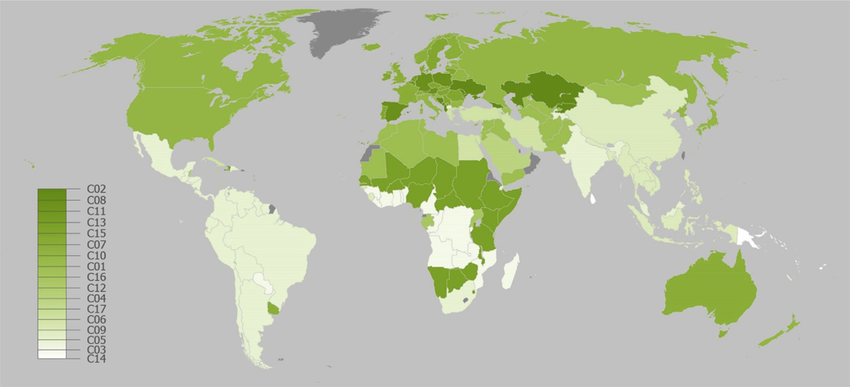
5. — A better option is to move to a leaner place altogether. If you’re in the United States, we recommend Colorado. Colorado is the leanest state, has exceptionally pure water sources, individual cities and counties in Colorado are extreme lean outliers, etc. Unbelievably, this comic exists:

If Colorado doesn’t suit you, you can move to some other state — Hawaii and Massachusetts are not far behind. To find your dream location, look at the CDC’s list of states, or one of the datasets of county-level data like this one or this one, and find a location with a lower rate of obesity than where you currently live. Or pick one of the places from the list of leanest communities in the US.
6. — This may not be extreme enough. After all, even Colorado is more than 20% obese. So a more radical version of the same idea is moving to a leaner country altogether.
If you live in the United States, the good news is that most countries are less obese than where you live now, even if you live in Colorado. Especially good choices seem to be Japan, South Korea, and Thailand, but there are many options — for the whole picture, check out the summary from Our World in Data.
But don’t just take our word for it, listen to these happy customers. Like this person who lost weight over five months in Vietnam, this person who moved to Vietnam and lost 112 pounds in ten months, this person who lost about 4kg (9lbs) after about two months in Japan (and similar stories in the comments), this person who lost 5lbs on a two-week trip to Japan, or this person who lost 10lbs during a two-week trip to Japan, despite not keeping up with their exercise regimen. Most of these people attribute their weight loss to eating less and walking more, but you’ll also notice that most of them say it was easy to eat less and walk more, and that many of them report being surprised at how much weight they lost and how easily they lost it.
We’ve also gotten a number of similar stories from commenters on the blog. First up is Julius, who said:
I currently live in Seattle but have moved around a lot. I’ve made 6 separate moves between places where I drank the tap water (mostly USA/UK/Hungary) and places I haven’t (South East Asia, India, Middle East). Whenever I’ve spent significant time in bottled water countries I lost weight (up to 50 lbs), and each time, save one 3 month stretch in Western Europe, I gained it back in tap water countries. I also lost weight for the first time in the States (20 lbs) this year around the time I switched to filtered water.
There’s also a similar story from Ross:
Very thought provoking and well researched piece. How about Japan? Very low rates of obesity. Similar issues with chemical residue. Anecdotally when I moved to Japan from the West I began to lose weight involuntarily, down to a BMI of 22. When I moved back to the West I regained weight. It’s a big rich country with plenty of processed, packaged food.
And a story from Tuck about their daughter:
Yes, my daughter is going to college in Japan. They have the “Freshmen 15 lbs” over there as well, except it’s the 15 lbs the foreigners lose when they go on a Japanese diet. Got a few panicked messages about “not having anything to wear”… LOL
So before you sign up for the gastric bypass, try spending a couple months in a lean country and see how it goes.
Studies
The question “what do we do about it” also includes the question “what research comes next?” Here’s what we’re thinking.
Correlational Studies
A lot of people’s first instincts when reading this work is to propose correlational studies. (We don’t necessarily mean a literal correlation, we just mean something that’s not a controlled experiment.) But we think that correlational studies are the wrong way to go at this point.
The first reason is statistical. We covered this in Part IV but it bears repeating. Because most of the modern variation in obesity is genetic, the apparent effect of any contaminant will be quite small, probably no larger than r = 0.50 and maybe a lot smaller. In any study we could run, the range of the variable would probably be restricted, and when the range of a variable is restricted, the correlation always ends up looking smaller than it really is. Some people have proposed we do animal studies for more control — but this is also a bad choice statistically, since the obesity effects in animals seem to be smaller than the effects for humans.
The combination of these problems means that any correlational study would be searching for a pretty small effect, and that means you would need a huge sample size to even have a good chance of finding a potential relationship. So “run a quick correlational study” starts looking like “find a way to fund and organize a study with 1,000 mice”. While we love mice, this seems like an awful lot of them. And even if we have enough statistical power that we have a 90% chance to detect a relationship, that still means we have a 10% chance of missing the relationship altogether. We don’t love those odds.

Second, A Chemical Hunger already documents a lot of correlational evidence for contaminants in general, and for a few contaminants in particular, especially lithium. If you already find this evidence compelling, it’s hard to imagine that one more piece of correlational evidence will do anything for you. And if you don’t find our review convincing, it’s hard to imagine that another piece of correlational evidence will change your mind.
The contamination theory of obesity has to be possible, in the sense that we know chemicals can cause weight gain and we know various chemicals are in the environment. We hope we’ve also convinced you that it’s plausible. Now we want to figure out, is it true? More correlational evidence isn’t going to get us there.
So overall we recommend going right for the jugular. If this theory is correct, then we have a good shot at doing what we really want to do — actually curing obesity — and no result could be more convincing than that.
Experiments
So in general, we approve of the idea of doing experiments to just cure obesity straight up.
Normally in public health it’s hard to do this kind of experiment, because it’s unethical to expose people to dangerous chemicals. Back when they were trying to figure out if cigarettes cause cancer, they didn’t do any studies where they assigned people to smoke 3 packs a day. But there’s nothing unethical about removing a contaminant from the environment, so we like that approach.
We call these experiments, and they are, but in many cases we can actually cheat a little by not bothering to include a control group. People almost never spontaneously stop being obese, so we can just use the general obesity rate in the population as our control group.
Generally speaking, there are two approaches. “Broad-spectrum” experiments take the overall contaminant theory seriously, and just try to reduce contaminant exposure generally, without committing to any specific contaminant. “Targeted” experiments go after one contaminant in particular, and see if controlling levels of that contaminant alone can lead to weight loss.
These have clear trade-offs. The broad-spectrum experiments are more likely to work and require less experimental control, but if they cure obesity, they don’t tell us what contaminant is responsible (curing obesity would still be pretty cool tho). The targeted experiments are less likely to work because we might go after the wrong contaminant, or we might fuck up our experimental control and let some contamination through — but if they DO work, then we have strong evidence that we’ve found the contaminant that’s responsible.
For all of these studies, the big hurdle is that we don’t know how quickly obesity can be reversed, even under the best circumstances. It might also vary a lot for different people — we have no idea. So if we try any of these experiments, we need to run them for several months at the very least, just to get a good idea of whether or not it’s working. Maybe if we’re lucky we’ll find out you can cure obesity in 2 weeks; but 3 months, 6 months, or even 1 year seems more plausible.
Below, we propose a few basic ideas for experiments. These aren’t exhaustive — as we do more research, we may come up with new and better ways to try to cure obesity. But they seem like an ok place to start.
Broad-Spectrum Experiments
Slime Mold Time Mold’s Excellent Adventure
The idea is simple. Some places, like Colorado, are pretty lean relative to everywhere else. We think that’s because those places are less contaminated. So we find some people who are obese, and pay for them all to take a year-long vacation to Boulder, Colorado, and see if they lose any weight.

For better effect, go a step further and send them to one of the leanest countries in the world instead. Vietnam seems to be the leanest country in the world right now, at only about 2% obese, and rent is pretty cheap there, so that would be a good option. If you want to stay in heavily industrialized nations, Japan is a good alternative; if you want to stay in the English-speaking world, maybe the Philippines. There are lots of good places to choose from.
For full effect, you would want your participants to eat the local food and drink the local water as much as possible. If they’re eating American food and drinking American beer, then you’re right back where you started.
(If you know of any study abroad or similar programs that we could piggyback on, please let us know!)
Throw Water Filters at the Problem and See What Happens
This is a broad-spectrum version of a targeted idea, below. The basic idea is simple. Contaminants might be in the water supply; filters get lots of stuff out of water; people drink water. So in this study, we find a bunch of people who are overweight or obese, send them the strongest/best water filters we can afford, and see if they lose any weight over the next several months.
For even more effect, send the filters to people who live in the most obese states, or even target some of the most obese communities directly.
This really is not a precision instrument — filters don’t get everything out of water, and water might not even be your main source of contaminants. Maybe your food or your carpets are the bigger problem. But if losing weight were as simple as throwing a water filter at the problem, that would be pretty exciting, and we would want to know.
Targeted Experiments
Right now lithium is our top suspect, so we’re using lithium as our go-to example in all of these experiments. But if it turns out that lithium isn’t a good match, any of these experiments could be retrofitted to target some other contaminant instead.
To use a targeted approach, we need to be able to figure out how much exposure people are getting, and we need to know what we can do to reduce that exposure. So there are a few pre-experiment projects we need to do first.
To begin with, we need to figure out which water filters (if any!) remove lithium from drinking water. If we can find a filter that works, this will let us make sure any water source is lithium-free.
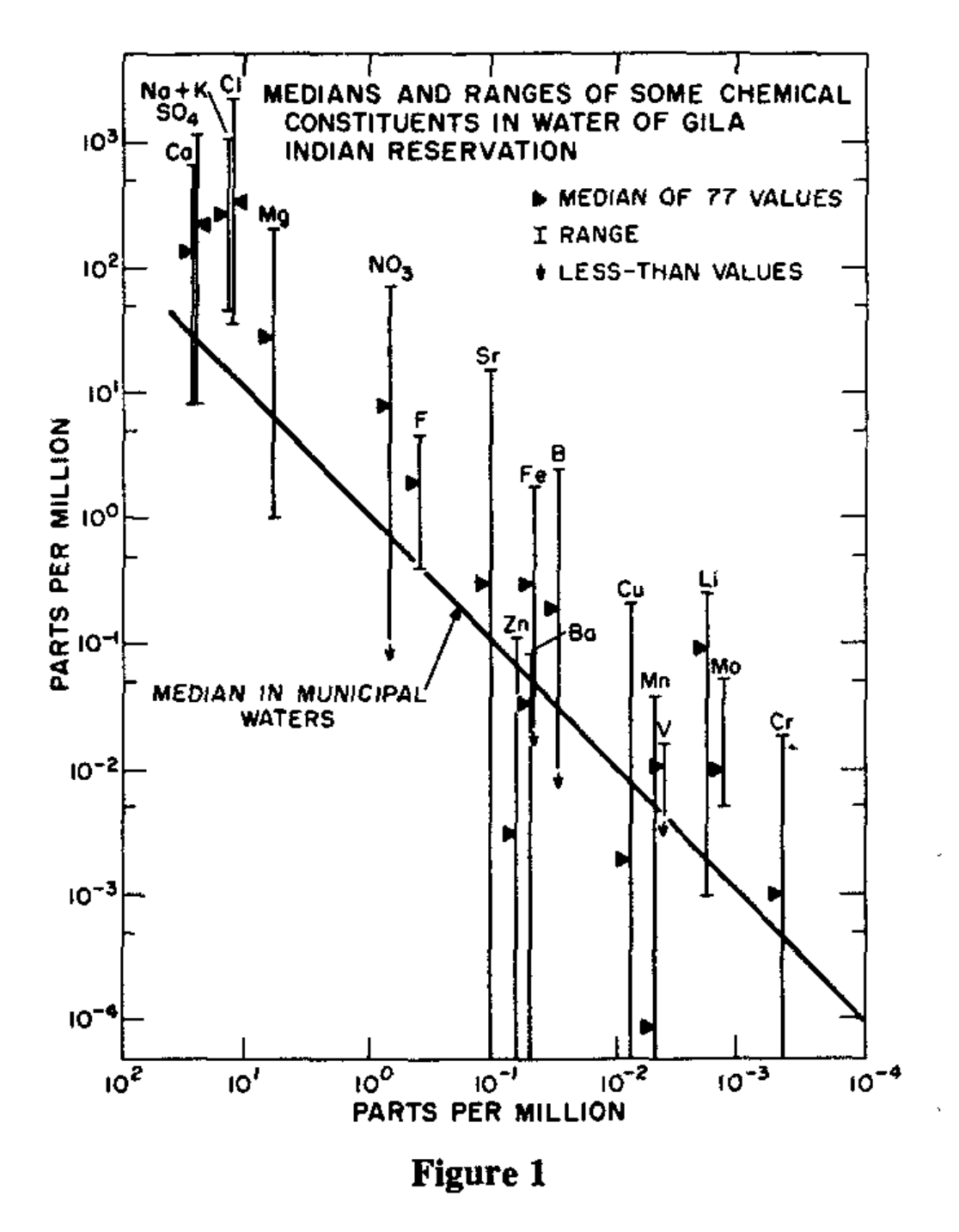
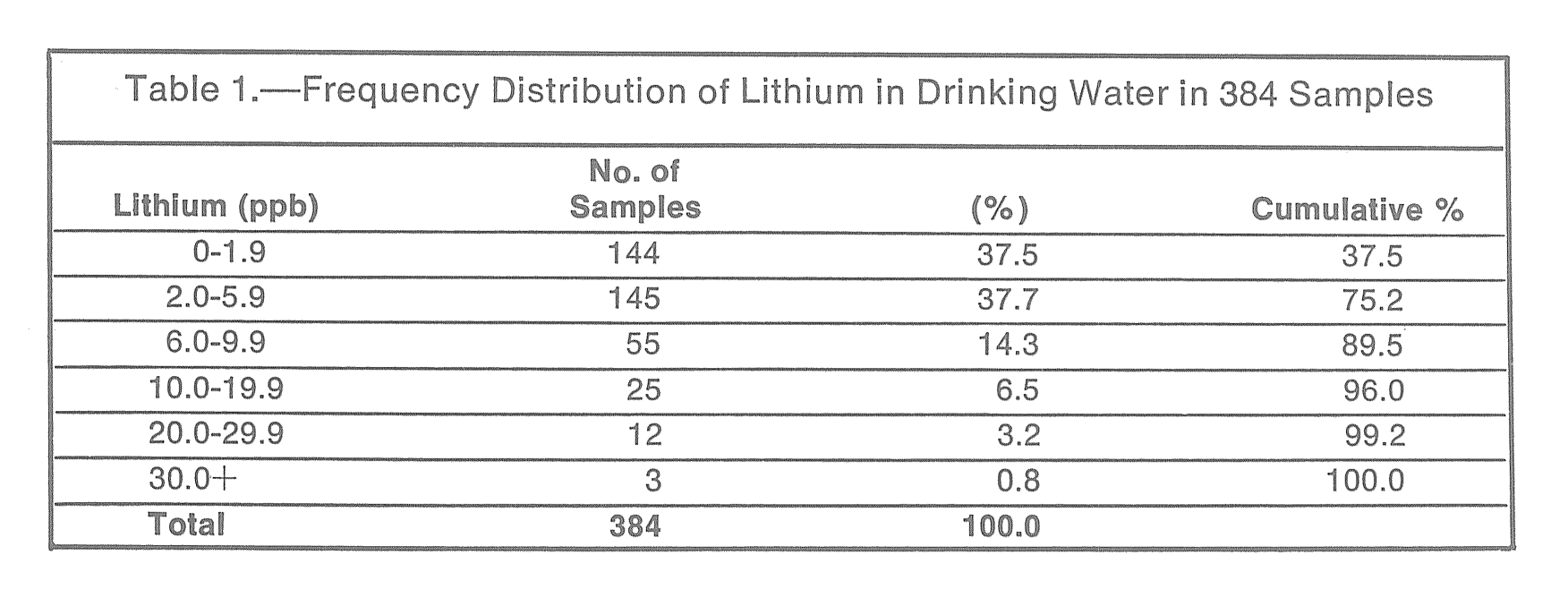
In addition, we’re worried that lithium might accumulate in food, so we need to do another study where we look at as many different types of food as we can and try to figure out if there are high levels of lithium in any of the stuff we’re all eating. Without this, any study will be hopelessly complicated because we won’t be able to control for the lithium in your food. But if we figure out what crops (if any) are concentrating lithium, maybe we can figure out a way to feed people a low-lithium diet.
Targeted Water Filters
Assuming we can find a water filter that does the job, we could do a pretty straightforward study where we send people a water filter that takes lithium out of their water, and see if they lose weight over a couple months.
For maximum effect, we would also want to make sure they weren’t getting any lithium from their food, which is why we want to do a study on how much lithium is in the food supply. It’s not clear how easy this would be — we might have to curate food sources and provide people with all their meals as well, which would make this study a hundred times more complicated.

There are a couple other things we could do to improve this study. We could focus on sending water filters to people in the most obese parts of the country, or to places where we already know the water is contaminated with lithium.
We could test the amount of lithium in people’s blood, urine, and/or saliva as they use the filter, see if it goes down, and see if the decrease in lithium in their body tracks on to weight loss. Assuming people did lose weight, this would be important because it might help us figure out more about the mechanism of lithium leaving the body. Some people will probably clear lithium faster than others, and if lithium causes obesity, we would want to be able to figure out how to help people clear it from their body as fast as possible.
We could also do a slightly bigger study, where we go to one of the fattest places in the US and install a bunch of whole-home water filtration systems for a couple randomly selected families who are overweight or obese. This would be more expensive but it would have some perks. If it turns out that showering in lithium-tainted water is really the active ingredient, and not just drinking it, then a whole-home water filtration system would take care of that.
There’s also a small chance that there’s just no filter on the market that can get lithium out of drinking water. Or maybe distillation works, but the cost is prohibitive for a whole-home system. In that case, we could rent a few water tanker trucks, fill them with water we know is low in lithium (we’ll import it from Colorado if we have to!), and take them to a cul-de-sac in one of the most obese communities in the US. If we can find a neighborhood who’d sign up for this, we could switch their houses’ water supplies over to our tanker trucks for a few months, bringing in new water as needed, and see if that did anything for their health.
Amish Obesity
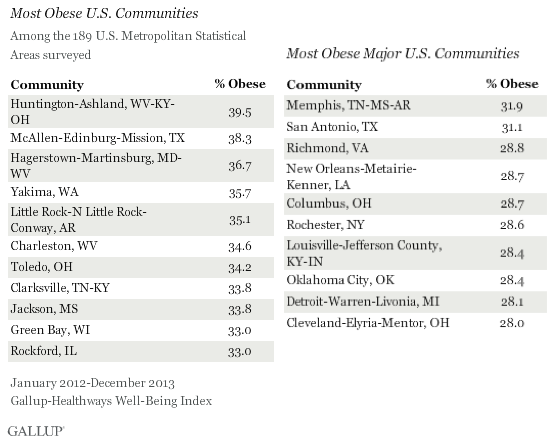
This piece from the LA Times is pretty bad, but it tells an interesting story. In part of Ontario, Canada, a group of Old Order Amish have “stunningly low obesity levels, despite a diet high in fat, calories and refined sugar.” The figure they quote is an obesity rate of only 4%. But about 200 miles south, the Amish in Holmes County, Ohio have obesity rates similar to the rest of the population, closer to 30% obese.
These two groups should be genetically similar. Both groups grow most of their own food. Both of them have pretty similar lifestyles — despite what the LA Times piece and this related article say, even if “only” 40% of the Amish in Ohio do hard farm labor, their lives are still more like the Amish in Ontario than the non-Amish in Holmes Country.
This makes them almost a perfect comparison. Why are the Amish in Ohio so much more obese than the Amish in Ontario? If the contamination hypothesis is correct, then we should be able to look at the local environments of these two communities and find more contamination (of one sort or another) in Ohio than in Ontario.
Because both groups grow most of their own food (we think?), we don’t need to worry about the influence of food imported from elsewhere — whatever contaminants are in their water will also be in their plants, and they won’t be bringing in contaminated food from outside. This makes this situation a much more controlled environment to study our hypothesis.

If lithium is the contaminant that causes obesity, we might expect to see deeper wells in Ohio than in Ontario. Information about the Amish is hard to find on the internet, for obvious reasons, but we have found some information that suggests that the Amish in America do use drilled wells, some of which may be relatively recent. We can’t find anything about the wells used by the Amish in Ontario — but it would be interesting if they were still using older, shallower wells for their water.
Another thing we might expect to see, if lithium is to blame, is evidence of some kind of fossil fuel activity in Ohio and not in Ontario. Well, in our last post we did review evidence for fossil fuel contamination in a number of places in Ohio. And when we were looking for documentation on water wells in Amish Ohio, we came across articles like Fracking on Amish Land (in Ohio), Energy Companies Take Advantage of the Amish Prohibition on Lawsuits (in Ohio), this excerpt about natural gas wells (in Pennsylvania), and Tradition, temptation as Amish debate fracking (in Pennsylvania, but mostly in Ohio).
Ontario has its own problems, including thousands of abandoned gas wells, but very few of them appear to be on Amish land. Zoom in on the towns of Milverton, Millbank, Newton, Linwood, and Atwood on that map, and you’ll see that there are almost no petroleum wells around these Amish communities. And unlike in Ohio, we haven’t found any news stories about recent drilling or fracking on Amish land in Ontario.
Or we could just go test the water. It’s a simple question, how much lithium is in the water in each place, and testing for other contaminants might not be a bad idea either. If we find similar levels of lithium in both places, and there are no complicating factors like imported food, that would be a strike against lithium as an explanation. But if there’s more lithium in the food and water in Ohio than in Ontario, that would be quite a mark in favor of the lithium hypothesis. Assuming they were interested, we could then work with the Amish in Ohio to try to get the lithium (or whatever) out of their water, and see if that reduced their rates of obesity.
We don’t expect that we have many Amish readers, but if you know of a good way to get in contact with the Amish in either of these locations, we’d be interested in talking to them!
Research Advising
There are also a few ideas we have that we won’t be pursuing ourselves, but if someone else (or a small team) wants to go after them, we would be happy to advise.
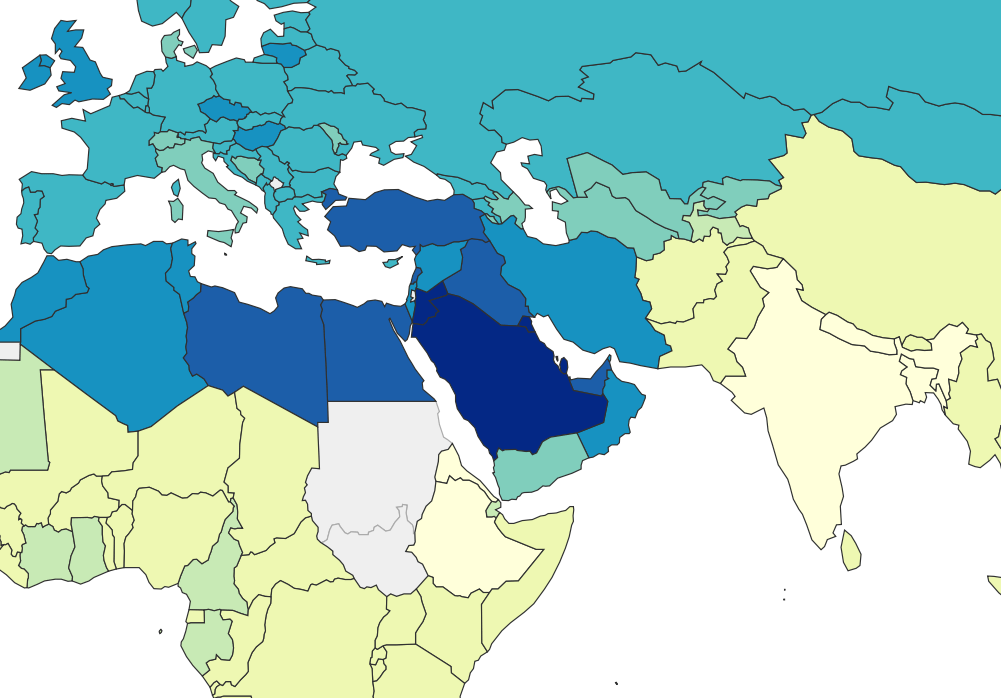
Taking lithium out of the water supply as a whole would be pretty hard, so it’s not usually an option. But it might be an option for countries that get most of their drinking water from desalination. You could run this as an experiment — one desalination plant uses lithium-free brine while another continues with the normal procedure — but you wouldn’t have to. In this case, there’s no need for a control group. If Saudi Arabia or Kuwait changed their desalination process so that no lithium ended up in their water, and saw their obesity rate fall 10% over the next five years, that would be evidence enough. Or you could do a version of this study with some other relevant group, e.g. seafarers drinking desalinated water as suggested by commenter ugoglen. So if anyone is able to do something like this, we would be interested in being involved.

In our post on PFAS, we did a small amount of regression modeling using data from The National Health and Nutrition Examination Survey (NHANES) and found evidence of a relationship between BMI and certain PFAS in the data for 1999-200, 2003-2004, and 2005-2006. This finding is very suggestive, but we only tested some very simple models, and we only looked at three of the datasets that are available. We think that a bigger analysis could be very illuminating, but model fitting isn’t our specialty. We would love to work with a data scientist or statistician with more model fitting experience, however, to conduct a more complete analysis. So if you have those skills and you’re interested, please let us know!
We’re still pretty interested in the all-potato diet. So far all we have are anecdotes, but the anecdotes are pretty compelling. Chris Voigt famously vowed to eat nothing but 20 plain potatoes (and a small amount of cooking oil) and lost 21 pounds over 60 days, without feeling very hungry. There’s also Andrew Taylor of Australia, who lost 114 lbs over a year of eating nothing but potatoes and reports feeling “totally amazing”. Last we heard he’s still doing pretty well. Magician Penn Jillette lost over 100 lbs using a strategy that started with two weeks of a potato-only diet (h/t reader pie_flavor), and seems to be keeping it off. This also inspired at least one copycat attempt from a couple who have jointly lost over 220 lbs starting with two weeks of an all-potato diet.

There’s also this comment from u/DovesOfWar on reddit:
To complement the potatoes anecdote, at some point to save money and time I ate almost nothing but potatoes, onions and butter and I lost like 60 pounds. I stopped because everyone thought I was starving (despite not being hungry) and I chugged it off to extreme lazyness/depression (despite not being sad) so I stopped doing that and never connected it to my diet, but what I should have done is write a fad book on the diet and solve the money problem that way. I’m back to a normal healthy 29 BMI now and still relatively poor, so I see I interpreted the experiment completely wrong and now my life sucks.
Based on those examples, you can see why we’re interested. It seems pretty low-cost (potatoes are cheap) and low-risk (if you feel bad, you can stop eating potatoes). If someone wants to organize a potato-centered weight-loss study, or if people just want to get together and try it for themselves, we’d be happy to advise. You can coordinate on the subreddit u/pondgrass set up over at r/spudbud if you like, though so far there doesn’t seem to be much activity.

We’re also interested in the effect of alkali metal ions, especially potassium. Lithium, currently our prime suspect, is an alkali metal ion that appears to affect the brain. Other alkali metal ions like sodium and potassium also play an important role in the brain, and there’s evidence that these ions may compete with each other, or at least interact, in interesting ways (see also here, here, and here). If lithium causes obesity, it may do so by messing with sodium or potassium signaling (or maybe calcium) in the brain, so changing the amount of these ions you consume, or their ratios, might help stop it.
This is supported by some hints that potassium consumption is related to successful weight loss. Potatoes are high in potassium, so if the all-potato diet really does work, that might be part of the mechanism.
You can easily get sodium from table salt, and you can get potassium from potassium salts like this one or this one. We’ve tried them, and we find them a little gross, but to some people they taste just like regular salt. If that’s no good, there are always dietary sources like potatoes.
So trying various forms of alkali-metal diets — high-K+, high-K+/low-Na+, high-K+/high-Na+, high-K+/low-Ca2+, etc. — seems pretty easy and might prove interesting. As before, if someone wants to organize a community study around this angle, or if people want to try it for themselves, we’d be happy to advise. These salts are pretty safe, and not prescription medications, but they’re not quite as basic as potatoes — before you try seriously changing your sodium or potassium intake, please talk with your doctor.
Also, how about lithium grease? These greases are basically the perfect slow-release form of lithium, which make them kind of concerning. Mechanics work with lithium grease and are relatively obese. But there are alternative kinds of greases that don’t use lithium, and sometimes companies intentionally switch what kind of grease they use. If a company switched out lithium grease for some other grease in one of their factories, we could compare the weights of workers at that factory to workers at other factories, and see if there was any weight loss over the next few years. And what happens when mechanics who use lithium grease every day switch to a new job? What happens if they get promoted to a desk job? What happens when they retire? If you know a group of mechanics or some other group that works with lithium grease and might be interested, please let us know!
We’re also interested in advising original ideas. We love it when you send us ideas we never would have come up with ourselves. So if you have some great idea — a review of a contaminant we didn’t cover, another idea for a related study, relevant anecdotes that might inspire something, etc. — let us know. If we like it, we’ll do what we can to help — advise you, promote it, try to help you get funding, whatever.
This is the end of A Chemical Hunger. We will still write more about obesity, and probably more about contamination, but this is the end of the series. Thank you for reading, commenting, sharing, contributing, questioning, challenging, and yes, even disputing! We’ve learned a lot from your comments and questions — and we hope you’ve learned something from reading!
Even if you still don’t find our hypothesis convincing, thank you for reading the series all the way to the end! We think it’s great that you were willing to give our wacky idea the time of day. This kind of exploration is essential, even if some of the theories turn out to be a little silly. And even if our theory is totally wrong, someday someone will figure out the answer to this thing, and we’ll send the global obesity rate back down to 2%.
As we mentioned, we want to conduct some research to follow up on the book-length literature review you just finished reading. Our near-term goal is to better understand how people get exposed to contaminants, especially lithium, so we can give advice on how to avoid exposure. Our medium-term goal is to figure out what causes obesity, probably by trying to cure it in a sample population. Our long-term goal is to try to cure it everywhere. That would be pretty cool.
If you’re interested in supporting this research, you can become a patron on patreon, or contact us if you want to help fund a larger project.
In conclusion: Be excellent to each other. Party on, dudes.